Note on Accessibility: Persons using mobile devices may find some tables are not fully accessible. Note that you can view tables on a larger screen or in the PDF version of the report/monograph. If you need additional assistance, email us or use our contact form and identify the tables for which access is required. We will assist you in accessing the content. NIH has helpful information on accessibility.
Technical Report 601
NTP Technical Report on the Toxicology and Carcinogenesis Studies of Di(2-ethylhexyl) Phthalate (CASRN 117-81-7) Administered in Feed to Sprague Dawley (Hsd:Sprague Dawley SD) Rats

Abstract
Di(2-ethylhexyl) phthalate (DEHP) is a member of the phthalate ester chemical class that occurs commonly in the environment and to which humans are widely exposed. Lifetime exposure to DEHP is likely to occur, including during the in utero and early postnatal windows of development. To date, no carcinogenicity assessments of DEHP have used a lifetime exposure paradigm that includes the perinatal period (gestation and lactation). The National Toxicology Program (NTP) tested the hypothesis that exposure during the perinatal period would alter the DEHP carcinogenic response quantitatively (more neoplasms) or qualitatively (different neoplasm types).
Two chronic carcinogenicity assessments of DEHP were conducted in which Sprague Dawley (Hsd:Sprague Dawley SD) rats were exposed to dosed feed containing 0, 300, 1,000, 3,000, or 10,000 ppm DEHP for 2 years using different exposure paradigms. In Study 1, groups of 45 F0 time-mated females were provided dosed feed beginning on gestation day (GD) 6 through lactation. On postnatal day (PND) 21, groups of 50 F1 rats per sex continued on the study and were provided dosed feed containing the same DEHP concentration as their respective dam for 2 years. In Study 2, groups of 50 rats per sex, aged 6 to 7 weeks at study start, were provided dosed feed containing DEHP for 2 years.
Perinatal and Postweaning Study in Rats (Study 1)
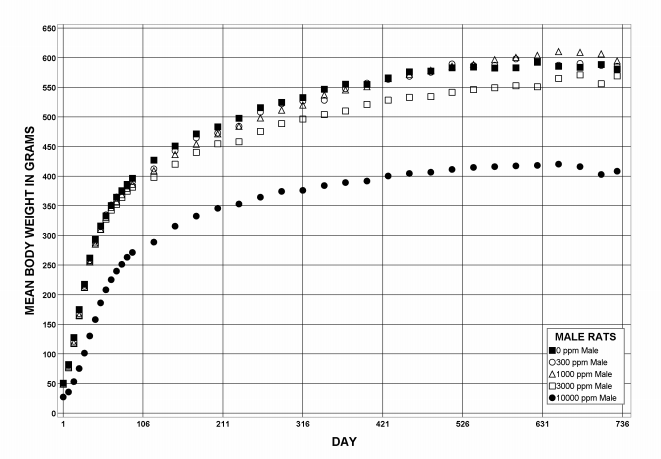
During the perinatal period, lower maternal mean body weight, maternal mean body weight gain, and feed consumption were observed in F0 dams exposed to 10,000 ppm DEHP relative to control animals. Also in that exposure group, litter size and pup weights on PND 1 were significantly decreased compared to the control group. Male and female pup mean body weight gains were significantly decreased in the 10,000 ppm group during lactation and resulted in significantly decreased pup body weights at weaning when compared to the control group. Pup survival was not affected following gestational and lactational DEHP exposure.
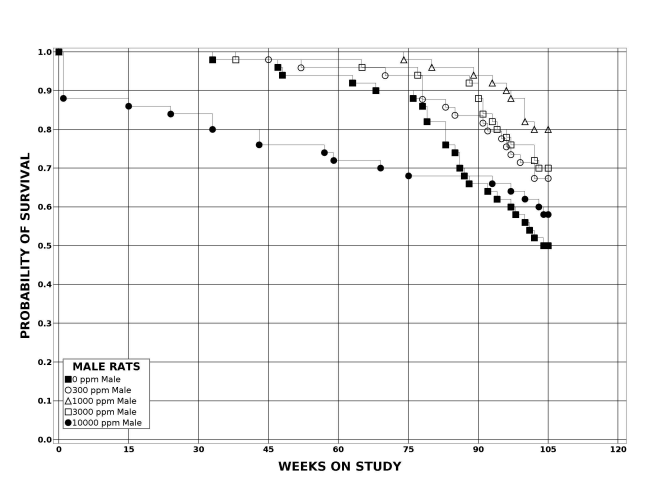
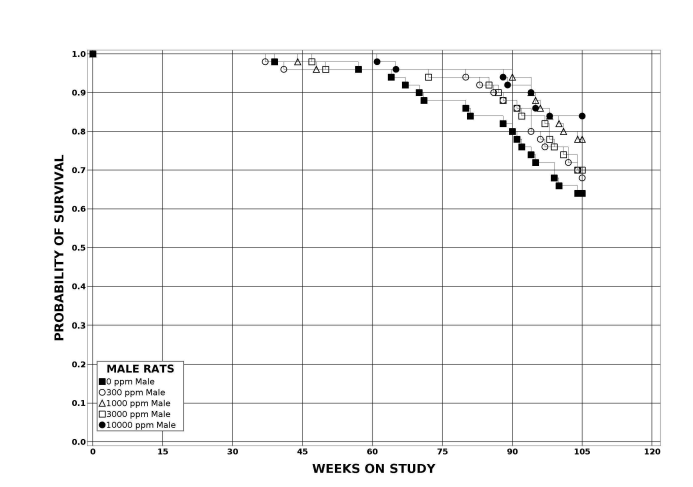
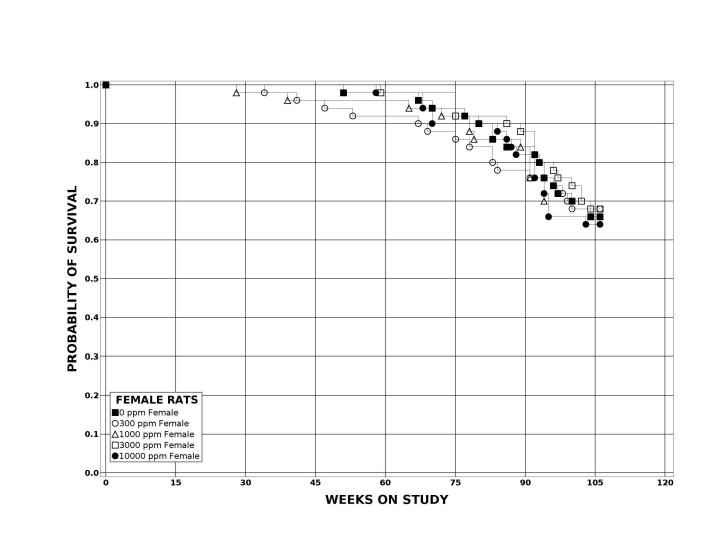
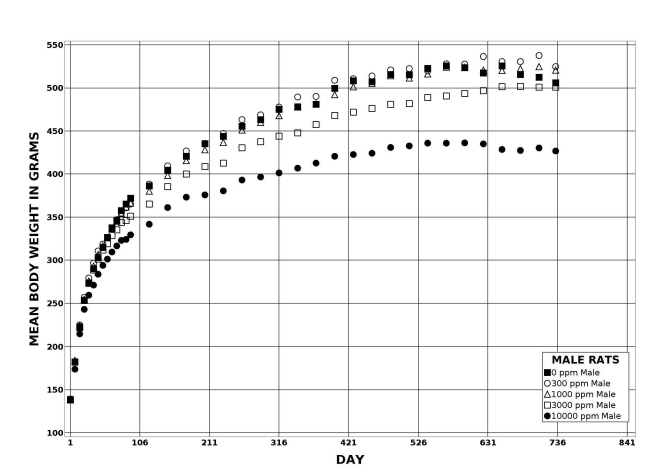
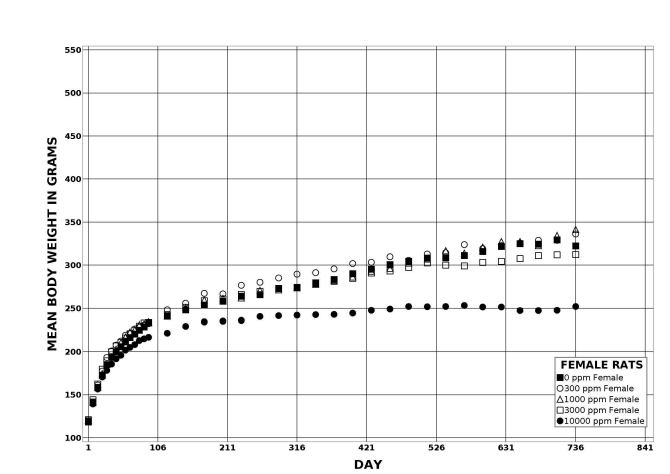
Following perinatal and 2 years of postweaning DEHP exposure, survival of exposed male and female rats to study termination was similar to that of control groups; however, there were decreases in mean body weight in the 10,000 ppm group compared to the control group.
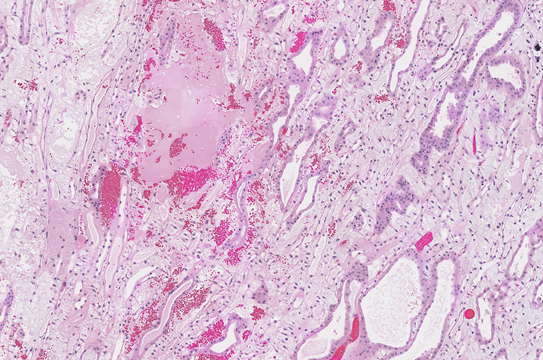
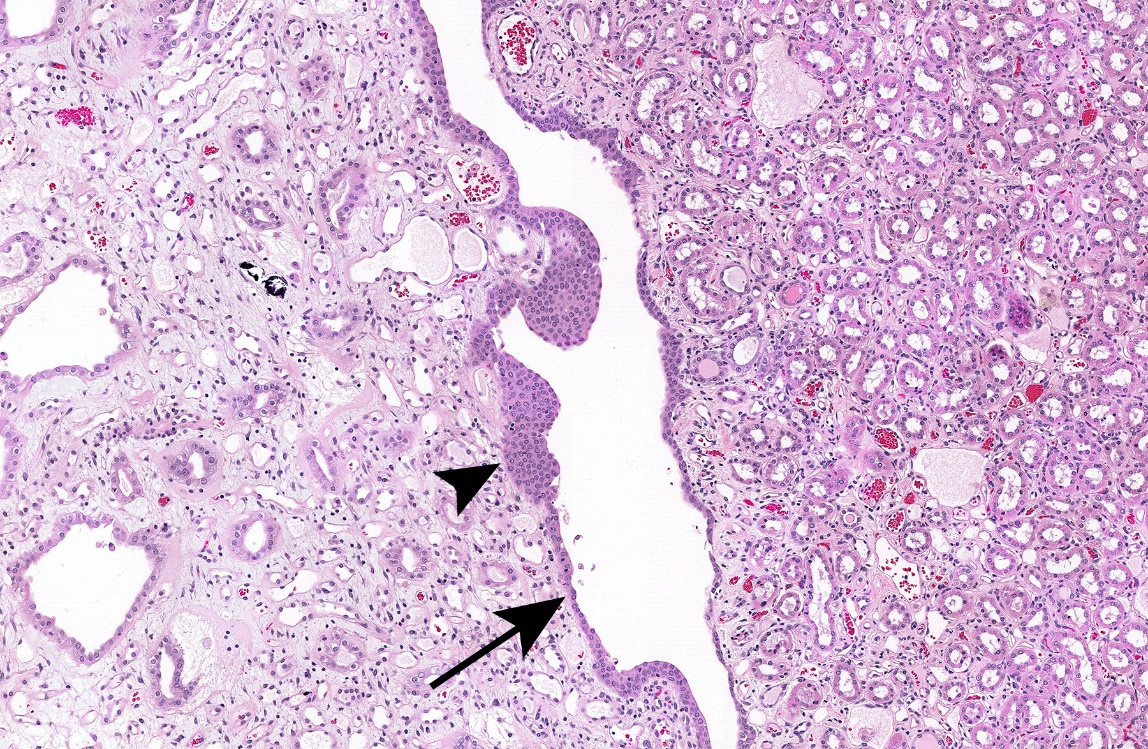
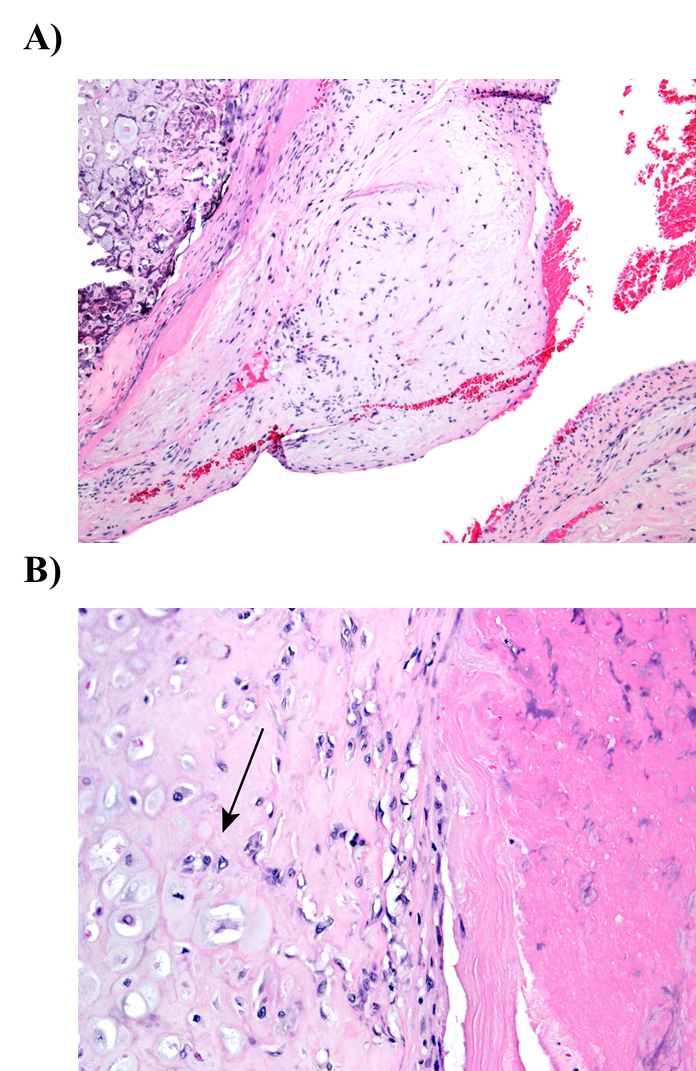
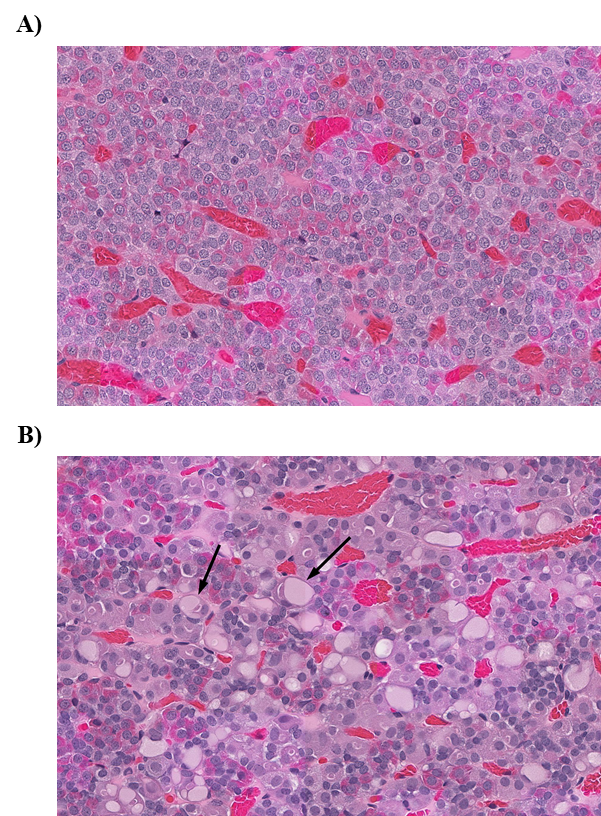
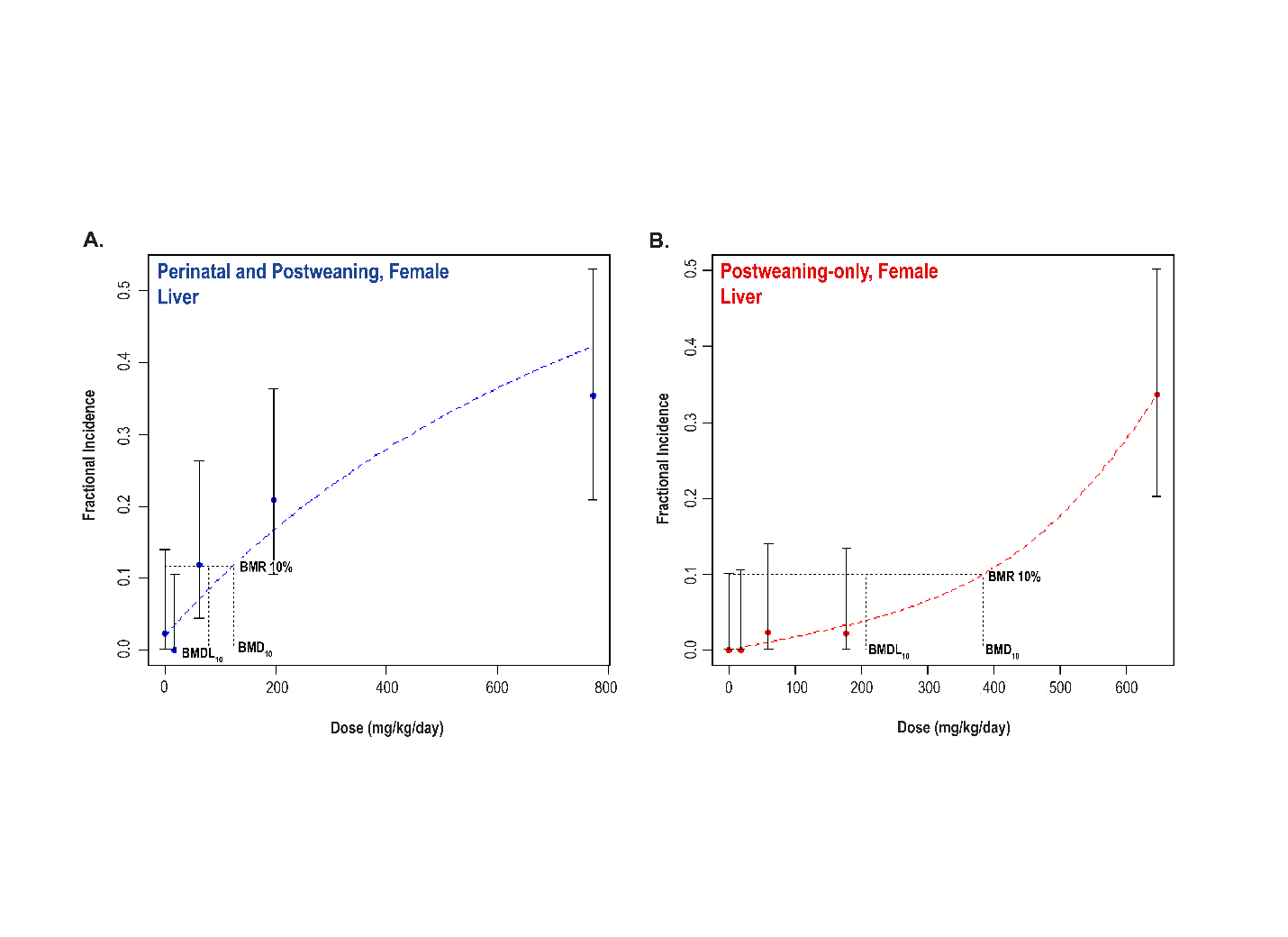
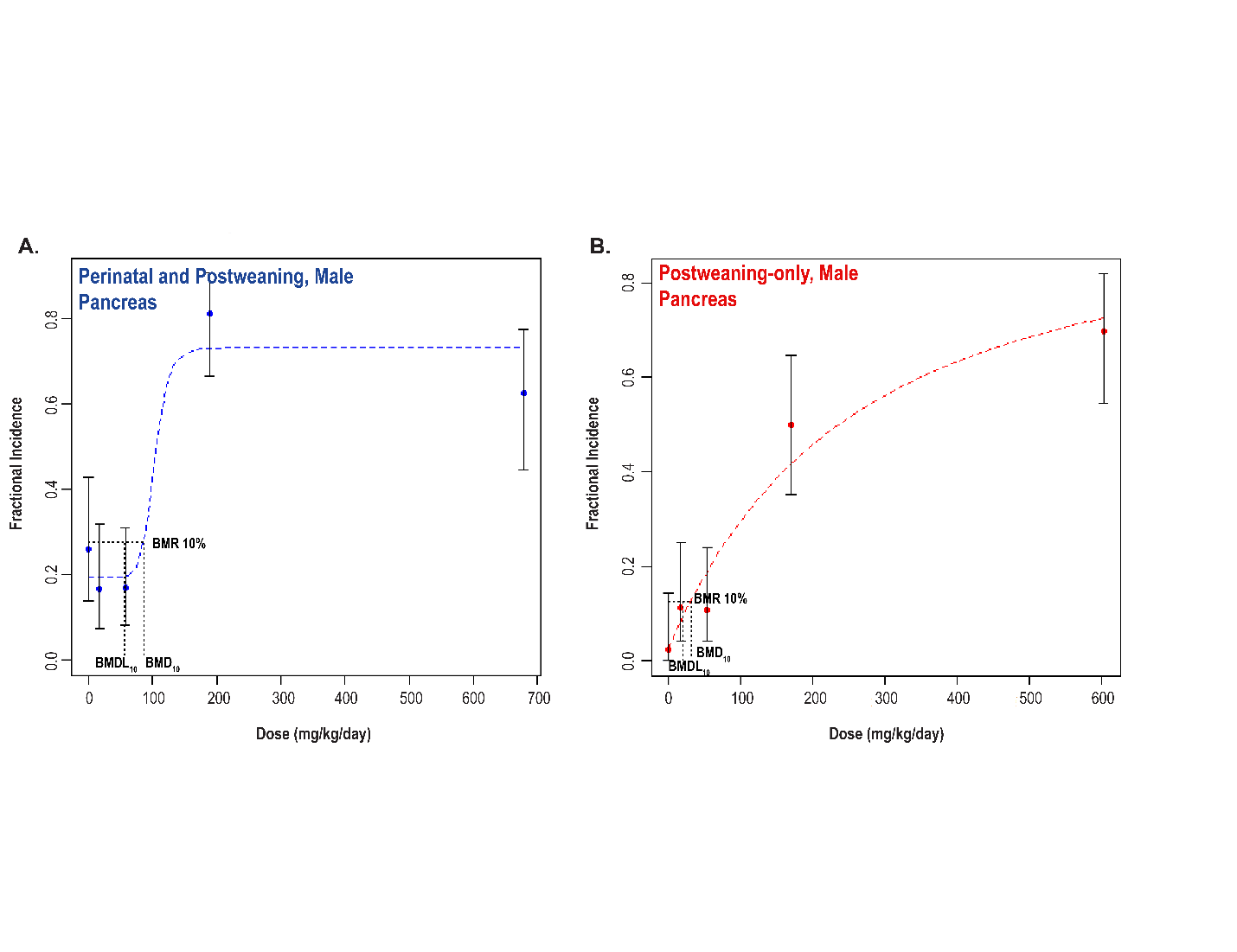
Significant increases in the incidences of hepatocellular adenoma, hepatocellular adenoma or carcinoma (combined), pancreatic acinar adenoma, and pancreatic acinar adenoma or carcinoma (combined) were observed in the 3,000 and 10,000 ppm male rats relative to the control group. Higher incidences of hepatocellular carcinomas (10,000 ppm males) and pancreatic acinar carcinomas (3,000 ppm males) were also observed. In female rats, significant increases in the incidences of liver neoplasms occurred in the 3,000 ppm (hepatocellular adenoma and hepatocellular adenoma or carcinoma [combined]) and 10,000 ppm (hepatocellular carcinoma and hepatocellular adenoma or carcinoma [combined]) groups. Occurrences of pancreatic acinar adenomas were observed in the 3,000 and 10,000 ppm female groups, and a trend of higher incidence of uterine adenocarcinomas with increasing exposure was observed given the incidence in the 10,000 ppm group. Nonneoplastic lesions were observed in the liver (male and female), pancreas (female), testis, epididymis, kidney (male and female), heart (male only), bone marrow (male only), and pituitary gland (male only).
Postweaning-only Study in Rats (Study 2)
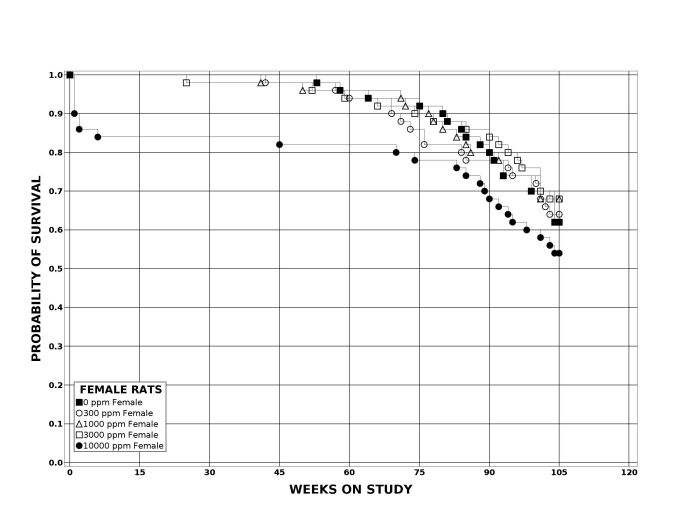
Following 2 years of postweaning DEHP exposure, survival of male and female rats was commensurate with or greater than that of control animals, and lower body weights were observed in the 10,000 ppm group. Notably, the magnitude of decreased weight was smaller in the control animals in Study 2 than in the control animals in Study 1. Significant increases in the incidences of hepatocellular adenoma, carcinoma, and adenoma or carcinoma (combined) were observed in male and female rats exposed to 10,000 ppm DEHP relative to the respective control group. In male rats, significantly increased incidences of pancreatic acinar neoplasms were observed in the 3,000 (adenoma) and 10,000 ppm (adenoma and carcinomas) groups. A trend of increasing incidence of testicular interstitial cell adenoma with increasing exposure was observed in male rats given the incidence observed in the 10,000 ppm DEHP group. In female rats, significantly increased incidences of uterine adenocarcinoma and uterine adenoma, adenocarcinoma, squamous cell carcinoma, or squamous cell papilloma (combined) were observed in the 10,000 ppm group compared to the control group. Occurrences of uterine squamous cell papilloma (including multiple) were observed in the 10,000 ppm group. Nonneoplastic lesions were observed in the liver (male and female), pancreas (male and female), testis, epididymis, uterus, heart (male only), bone marrow (male), and pituitary gland (male only).
Comparative Carcinogenic Benchmark Dose Analyses
Benchmark dose (BMD) levels corresponding to a 10% increased risk of carcinogenic response (BMD10) were estimated for exposure-related carcinogenic responses that were observed in both studies. Generally, the BMDs between studies were within threefold of each other. The lowest estimated BMD10 (30.99 mg DEHP/kg body weight/day) corresponded to pancreatic acinar adenoma or carcinoma (combined) in males in the postweaning-only study (Study 2).
Genetic Toxicology
DEHP was tested in a variety of genotoxicity assays in vitro and in vivo; most results were negative. In vitro, negative results were obtained in the following assays: six independent bacterial mutation assays in Salmonella typhimurium bacterial strains (TA100, TA1535, TA1537, TA97, and TA98) with and without exogenous metabolic activation systems (S9 mix; induced hamster, rat, and mouse liver S9), a single mouse lymphoma gene mutation assay (with and without induced rat liver S9 mix), and three independent chromosomal aberration assays conducted in Chinese hamster ovary (CHO) cells (with and without rat liver S9). In nine in vitro sister chromatid exchange tests conducted in CHO cells with and without S9, DEHP produced positive responses in four tests, equivocal results in three, and negative results in two.
In vivo, no increases in chromosomal aberrations were observed in bone marrow cells of female B6C3F1 mice following exposure to DEHP in dosed feed for 14 days. DEHP produced mixed results in three independent erythrocyte micronucleus assays: equivocal in female B6C3F1 mice exposed to DEHP in dosed feed for 14 days, equivocal in male TgAC (FVB/N) mice and positive in female TgAC (FVB/N) mice following exposure via dosed feed for 26 weeks, and negative in male and female TgAC (FVB/N) mice following a 26-week dermal exposure. DEHP produced negative results in two independent studies that tested for induction of sex-linked recessive lethal mutations in Drosophila melanogaster.
Conclusions
Under the conditions of the perinatal and postweaning feed study (Study 1), there was clear evidence of carcinogenic activity of di(2-ethylhexyl) phthalate (DEHP) in male Hsd:Sprague Dawley SD rats based on the increased incidences of hepatocellular adenoma or carcinoma (combined) and acinar adenoma or carcinoma (combined) neoplasms (predominately adenomas) of the pancreas. There was clear evidence of carcinogenic activity of DEHP in female Hsd:Sprague Dawley SD rats based on the increased incidence of hepatocellular adenoma or carcinoma (combined). The occurrence of pancreatic acinar adenoma or carcinoma (combined) was considered to be related to exposure. The occurrence of uterine (including cervix) adenoma, adenocarcinoma, squamous cell carcinoma, or squamous cell papilloma (combined) in female rats may have been related to exposure.
Under the conditions of the postweaning-only feed study (Study 2), there was clear evidence of carcinogenic activity of DEHP in male Hsd:Sprague Dawley SD rats based on the increased incidences of hepatocellular adenoma or carcinoma (combined) and acinar adenoma or carcinoma (combined) neoplasms (predominately adenomas) of the pancreas. The occurrence of testicular interstitial cell adenoma in male rats may have been related to exposure. There was clear evidence of carcinogenic activity of DEHP in female Hsd:Sprague Dawley SD rats based on the increased incidences of hepatocellular adenoma or carcinoma (combined) and uterine (including cervix) adenoma, adenocarcinoma, squamous cell carcinoma, or squamous cell papilloma (combined). The occurrence of pancreatic acinar adenoma or carcinoma (combined) in female rats was considered to be related to exposure.
The BMD analysis shows there was no consistent pattern indicating that perinatal and postweaning exposure was more sensitive compared to postweaning-only exposure and modeled responses were within threefold of each other. However, there was a stronger carcinogenic response in the reproductive organs (uterus and testis) in the postweaning-only exposure study compared to the perinatal and postweaning exposure study.
Perinatal and postweaning exposure to DEHP (Study 1) resulted in increased incidence of nonneoplastic lesions in the liver, kidney, heart (male), pancreas (female), pituitary gland (male), bone marrow (male), testis, and epididymis. In addition, exposure increased gross lesions within the reproductive tract of males and females.
Postweaning exposure to DEHP (Study 2) resulted in increased incidence of nonneoplastic lesions in the liver, pancreas, heart (male), pituitary gland (male), bone marrow (male), testis, epididymis, and uterus.
Synonyms: Bis(2-ethylhexyl)phthalate; dioctyl phthalate; phthalic acid di(2-ethylhexyl) ester; bis(2-ethylhexyl) 1,2-benzenedicarboxylate; 1,2-benzenedicarboxylic acid bis(2-ethylhexyl) ester
Trade names: Platinol DOP; Octoil; Silicol 150; Bisoflex 81; Eviplast 80
Summary of the Two-year Carcinogenesis and Genetic Toxicology Studies of Di(2-ethylhexyl) Phthalate
| Perinatal and Postweaning Study (Study 1) |
Postweaning-only Study (Study 2) |
|||
|---|---|---|---|---|
| Male Sprague Dawley Rats |
Female Sprague Dawley Rats |
Male Sprague Dawley Rats |
Female Sprague Dawley Rats |
|
| Concentrations in feed | 0, 300, 1,000, 3,000, or 10,000 ppm | 0, 300, 1,000, 3,000, or 10,000 ppm | 0, 300, 1,000, 3,000, or 10,000 ppm | 0, 300, 1,000, 3,000, or 10,000 ppm |
| Survival rates | 25/50, 33/49, 40/50, 35/50, 29/50 | 31/50, 32/50, 34/50, 34/50, 27/50 | 32/50, 35/50, 39/50, 35/50, 42/50 | 33/50, 34/50, 33/50, 34/50, 32/50 |
| Body weights | 10,000 ppm group 29.7% less than the control group | 3,000 ppm group 9.9% less than the control group; 10,000 ppm group 31.7% less than the control group | 10,000 ppm group 15.6% less than the control group | 10,000 ppm group 21.9% less than the control group |
| Gross lesions | Testis: small (2/49, 2/49, 4/50, 2/50, 45/49); enlarged (or swelling) (0/49, 0/49, 0/50, 0/50, 1/49); fluid or blood filled (0/49, 1/49, 1/50, 1/50, 1/49); right or left, abdominal, undescended (1/49, 0/49, 0/50, 0/50, 19/49); right or left, inguinal, undescended (0/49, 1/49, 1/50, 0/50, 4/49); right or left, abdominal or inguinal, undescended (1/49, 1/49, 1/50, 0/50, 23/49); right, not present (0/49, 0/49, 0/49, 0/50, 1/49); cranial suspensory ligament (0/49, 0/49, 0/50, 0/50, 5/49) Epididymis: small (0/49, 0/49, 2/50, 0/50, 14/49); right, cauda, agenesis (0/49, 0/49, 0/49, 0/50, 2/49); right or left, caput, agenesis (0/49, 0/49, 0/50, 0/50, 4/49); right or left, cauda, agenesis (0/49, 0/49, 0/50, 0/50, 2/49); right or left, corpus, agenesis (0/49, 0/49, 0/50, 0/50, 3/49) Levator ani/bulbocavernosus muscle: small (0/50, 0/49, 0/50, 0/50, 2/48) Cowper’s glands: left, small (0/50, 0/49, 0/50, 0/50, 1/47); right, small (0/50, 0/49, 0/50, 0/50, 1/47) Prostate glands: small (0/50, 0/49, 0/50, 0/50, 1/47) Seminal vesicles/ coagulating glands: small (1/50, 0/49, 1/50, 1/50, 8/47) Phallus: small (0/50, 0/49, 0/49, 0/50, 3/49); cleft (0/50, 0/49, 0/49, 0/50, 3/49) Prepuce: cleft (0/50, 0/49, 0/50, 0/50, 1/49); incomplete preputial separation (0/50, 0/49, 0/50, 0/50, 7/49) Gubernaculum: right or left, not present (0/47, 0/49, 0/49, 0/50, 18/41); ↑ right length |
Vagina: not patent (0/50, 0/50, 0/50, 0/50, 5/48) Phallus: cleft (0/50, 0/50, 0/50, 2/50, 1/48) |
None | None |
| Nonneoplastic effects | Liver: hepatocyte, cytoplasmic alteration (0/50, 0/49, 1/50, 28/50, 37/49); hepatocyte, hypertrophy (0/50, 0/49, 0/50, 3/50, 17/49); pigment (0/50, 1/49, 5/50, 40/50, 38/49); necrosis (3/50, 4/49, 1/50, 6/50, 13/49); eosinophilic focus (4/50, 1/49, 7/50, 2/50, 11/49); basophilic focus (1/50, 1/49, 4/50, 4/50, 17/49) Testis: germinal epithelium, degeneration (includes bilateral) (16/49, 25/49, 21/50, 21/50, 44/49); interstitial cell, hyperplasia, focal (includes bilateral) (4/49, 3/49, 6/50, 5/50, 30/49); seminiferous tubule, dysgenesis (includes bilateral) (0/49, 0/49, 0/50, 0/50, 10/49) Epididymis: hypospermia (includes bilateral) (4/49, 5/49, 12/50, 8/50, 43/49) Kidney: papilla, edema (0/50, 0/49, 0/50, 0/50, 39/49); papilla, hemorrhage (0/50, 1/49, 0/50, 2/50, 12/49); epithelium, papilla, hyperplasia (9/50, 4/49, 4/50, 3/50, 17/49); infarct (2/50, 10/49, 9/50, 7/50, 17/49) Heart: valve, fibrosis (0/50, 2/49, 1/50, 3/50, 11/49); valve, thrombus (0/50, 0/49, 0/50, 0/50, 6/49) Bone marrow: hypercellularity (21/50, 17/49, 29/50, 34/50, 36/50) Pituitary gland: pars distalis, hypertrophy (3/50, 7/49, 5/50, 15/50, 37/49) |
Liver: hepatocyte, cytoplasmic alteration (0/49, 4/50, 7/50, 39/50, 39/48); hepatocyte, hypertrophy (0/49, 2/50, 5/50, 9/50, 34/48); pigment (0/49, 6/50, 14/50, 36/50, 40/48); necrosis (3/49, 9/50, 3/50, 7/50, 8/48); eosinophilic focus (3/49, 4/50, 4/50, 7/50, 12/48); basophilic focus (4/49, 5/50, 3/50, 2/50, 10/48); bile duct hyperplasia (9/49, 13/50, 13/50, 21/50, 8/48) Pancreas: acinus, hyperplasia (0/49, 0/50, 0/50, 2/50, 3/48) Kidney: papilla, edema (0/50, 0/50, 2/50, 0/50, 38/48); epithelium, papilla, hyperplasia (2/50, 1/50, 2/50, 4/50, 15/48); infarct (0/50, 3/50, 7/50, 5/50, 12/48); renal tubule, cyst (0/50, 0/50, 2/50, 0/50, 7/48); renal tubule, dilation (0/50, 0/50, 0/50, 0/50, 3/48) |
Liver: hepatocyte, cytoplasmic alteration (0/50, 1/50, 0/50, 38/50, 49/50); hepatocyte, hypertrophy (0/50, 0/50, 0/50, 2/50, 6/50); pigment (0/50, 0/50, 7/50, 45/50, 50/50); necrosis (0/50, 2/50, 4/50, 7/50, 8/50); eosinophilic focus (1/50, 0/50, 4/50, 2/50, 24/50); clear cell focus (29/50, 31/50, 33/50, 35/50, 39/50) Pancreas: acinus, hyperplasia (7/49, 8/50, 9/50, 24/50, 26/50) Testis: germinal epithelium, degeneration (includes bilateral) (31/50, 25/50, 21/50, 22/50, 50/50); edema (includes bilateral) (27/50, 23/50, 29/50, 24/50, 45/50); interstitial cell, hyperplasia, focal (includes bilateral) (1/50, 1/50, 0/50, 4/50, 4/50) Epididymis: hypospermia (includes bilateral) (4/50, 4/50, 4/50, 3/50, 43/50); duct, exfoliated germ cell (includes bilateral) (2/50, 3/50, 4/50, 4/50, 36/50) Heart: valve, fibrosis (2/50, 0/50, 0/50, 1/50, 9/50); valve, thrombus (0/50, 0/50, 0/50, 2/50, 6/50) Bone marrow: hypercellularity (18/50, 22/50, 30/50, 25/50, 34/50) Pituitary gland: pars distalis, hypertrophy (8/50, 10/50, 11/50, 14/50, 37/50) |
Liver: hepatocyte, cytoplasmic alteration (0/50, 2/50, 15/50, 38/50, 45/49); hepatocyte, hypertrophy (0/50, 0/50, 6/50, 14/50, 28/49); pigment (3/50, 0/50, 18/50, 30/50, 48/49) Pancreas: acinus, hyperplasia (0/50, 1/50, 1/50, 1/50, 5/47) Uterus: inflammation, chronic (2/50, 9/50, 6/50, 8/50, 8/49) |
| Neoplastic effects | Liver: hepatocellular adenoma (0/50, 1/49, 0/50, 3/50, 8/49); hepatocellular carcinoma (1/50, 0/49, 0/50, 0/50, 3/49); hepatocellular adenoma or carcinoma (combined) (1/50, 1/49, 0/50, 3/50, 11/49) Pancreas: acinar adenoma (10/50, 7/49, 8/50, 36/50, 22/49); acinar carcinoma (0/50, 0/49, 0/50, 3/50, 1/49); acinar adenoma or carcinoma (combined) (10/50, 7/49, 8/50, 38/50, 22/49) |
Liver: hepatocellular adenoma (1/49, 0/50, 5/50, 9/50, 5/48); hepatocellular carcinoma (0/49, 0/50, 0/50, 0/50, 8/48); hepatocellular adenoma or carcinoma (combined) (1/49, 0/50, 5/50, 9/50, 13/48) Pancreas: acinar adenoma or carcinoma (combined) (0/49, 0/50, 0/50, 2/50, 1/48) |
Liver: hepatocellular adenoma (0/50, 2/50, 0/50, 1/50, 6/50); hepatocellular carcinoma (0/50, 0/50, 0/50, 0/50, 6/50); hepatocellular adenoma or carcinoma (combined) (0/50, 2/50, 0/50, 1/50, 12/50) Pancreas: acinar adenoma (1/49, 4/50, 5/50, 23/50, 30/50); acinar carcinoma (0/49, 1/50, 0/50, 1/50, 5/50); acinar adenoma or carcinoma (combined) (1/49, 5/50, 5/50, 23/50, 33/50) |
Liver: hepatocellular adenoma (0/50, 0/50, 1/50, 1/50, 13/49); hepatocellular carcinoma (0/50, 0/50, 0/50, 0/50, 2/49); hepatocellular adenoma or carcinoma (combined) (0/50, 0/50, 1/50, 1/50, 14/49) Pancreas: acinar adenoma or carcinoma (combined) (0/50, 0/50, 0/50, 1/50, 2/47) Uterus: adenoma, adenocarcinoma, squamous cell carcinoma, or squamous cell papilloma (combined) (2/50, 4/50, 1/50, 6/50, 13/50) |
| Equivocal findings | None | Uterus: adenoma, adenocarcinoma, squamous cell carcinoma, or squamous cell papilloma (combined) (3/50, 1/50, 1/50, 3/50, 7/50) | Testis: interstitial cell, adenoma (7/50, 3/50, 3/50, 6/50, 15/50) | None |
| Level of evidence of carcinogenic activity | Clear evidence | Clear evidence | Clear evidence | Clear evidence |
| Genetic toxicology Bacterial gene mutations: Negative in Salmonella typhimurium strains TA100, TA1535, TA1537, TA97, and TA98, with and without S9 Mouse lymphoma L5178Y tk+/− cells: Negative with and without S9 In vitro CHO cell chromosomal aberration test: Negative with and without S9 In vitro CHO cell sister chromatid exchange test: Without rat liver S9: Positive or equivocal in 7 out of 9 studies With rat liver S9: Negative in 9 out of 9 studies In vivo chromosome aberration test: Negative in female B6C3F1 mice exposed via dosed feed for 14 days In vivo micronucleus test in mice: B6C3F1 mice: Equivocal in females exposed via dosed feed for 14 days TgAC (FVB/N) mice: Equivocal in males and positive in females exposed via dosed feed for 26 weeks TgAC (FVB/N) mice: Negative in males and females exposed dermally for 26 weeks Drosophila melanogaster sex-linked recessive lethal test: Adult injection: Negative Larval feeding: Negative |
||||
Introduction
Chemical and Physical Properties
Di(2-ethylhexyl) phthalate (DEHP) is the diester of phthalic acid and the branched-chain 2-ethylhexanol. DEHP is a pale-yellow to colorless viscous liquid at room temperature and can have a slight odor. DEHP has a boiling point of 384°C, a melting point of −55°C, and a flash point of 215°C.1 At 25°C, DEHP has a limited water solubility of approximately 0.3 mg/L and a vapor pressure ranging from 1.42 × 10−7 to 9.75 × 10−6 mm Hg.2 DEHP has an estimated log Kow of 7.63 and is miscible in organic solvents, such as hexane.
Production, Use, and Human Exposure
DEHP is a widely used member of the phthalate ester chemical class. Phthalates are employed predominantly as plasticizers to provide flexibility in products composed of polyvinyl chloride (PVC) plastic or vinyl chloride resins. DEHP is produced by the esterification of phthalic anhydride with 2-ethylhexanol in the presence of an acid catalyst, such as sulfuric acid or para-toluenesulfonic acid.4 DEHP is considered a high-production volume chemical with an estimated 10 to 50 million pounds produced in the United States in 2015, as reported to the U.S. Environmental Protection Agency (EPA),5 a production level consistent with annual production reports from 1986 to 2014, indicating that DEHP use remained consistent.
Globally, between 90% and 95% of DEHP is used as a plasticizer in the manufacture of PVC polymers and corresponding products.6,7 DEHP is used in a variety of plastic consumer products, including construction materials, shower curtains, garden hoses, floor tiles, automobile upholstery, and food packaging materials. Plastics may contain 1% to 40% DEHP by weight, with materials that exhibit increased softness or flexibility likely containing higher levels of DEHP or other phthalates. DEHP is used in the production of medical devices, such as blood bags, enteral/parenteral nutrition bags, peritoneal dialysis bags, and medical tubing.8,9 Because DEHP is not covalently bonded to the PVC polymer, potential exists for DEHP to leach into contact media. Migration from PVC storage bags into collected blood, blood products, and other biological products is likely associated with the lipophilic nature of DEHP.
Exposure to DEHP can occur via numerous pathways, such as contact with DEHP-containing plastic products, consumption of foods packaged in plastics, drinking of well water near waste sites, workplace/indoor inhalation of aerosols or particulates containing DEHP, or exposure during certain medical procedures.4 The most common exposure pathway is through ingestion of food contaminated with DEHP, which typically occurs because of contact with plastic packaging materials. Migration efficiency of DEHP into foodstuffs from packaging materials is likely associated with the lipophilic nature of DEHP and the contact surface area with the packing materials. In the United States, average daily DEHP exposure from food is estimated to be 0.3 mg/day with a maximum of 2.0 mg/day per individual.10 Higher DEHP concentrations (≥300 μg/kg) have been noted in poultry, cooking oil, and cream-based dairy products relative to other assessed foodstuffs.11 In water, DEHP exhibits low solubility, suggesting a lower relative contribution of drinking water to estimated total daily exposure.12,13 Additionally, the low vapor pressure of DEHP indicates a limited capacity for DEHP to volatilize into the air; however, it can readily adsorb to dust particles that can then be respired or ingested. Fromm et al. measured concentrations of DEHP in indoor air and vacuum cleaner dust samples.14 The median indoor air DEHP concentration was 156 ng/m3 (95th percentile, 390 ng/m3) and 703.4 mg/kg (95th percentile, 1,542 mg/kg) in dust samples.
Measurable urinary DEHP metabolite concentrations from participants in the National Health and Nutrition Examination Survey (NHANES) indicate widespread exposure to DEHP in the U.S. population, but have been declining over the years.15 Urinary concentration (50th percentile) of a DEHP metabolite, mono(2-ethylhexyl) phthalate (MEHP) (2015–2016) was 1.24 μg/g of creatinine (95th percentile, 5.93 μg/g of creatinine).15 Using the NHANES data, researchers estimated a median cumulative DEHP exposure of 0.17 μg DEHP/kg body weight/day (μg/kg/day) (95th percentile, 12.0 μg/kg/day).16 Urinary concentrations of DEHP and its metabolites are higher in exposed workers relative to unexposed workers and are detected at higher concentrations in postshift relative to preshift samples.14,17,18 High exposures have been documented in workers in countries other than the United States and observed in various industries.19-22 Newborns and infants may be at risk for higher DEHP exposure relative to the general population due to differences in metabolic capacity, increased food, water, and air intake per unit body weight, and behaviors such as crawling and mouthing, which can increase exposure to contaminants present in soil, house dust, and consumer products.23 Additionally, DEHP and its metabolites have been detected in breast milk and baby formula. Average DEHP exposure in nursing infants has been estimated at between 6 and 24 µg/kg/day.24,25 Multiple DEHP metabolites have also been measured in human amniotic fluid samples, indicating exposure can occur in utero.26
DEHP exposure has been associated with certain medical procedures that use PVC plastic bags and tubing is thought to be much higher than from other anticipated environmental exposures. Parenteral exposure to DEHP can occur in patients undergoing medical procedures, such as intravenous administration of drugs, total parenteral nutrition, transfusion of blood or blood products, cardiopulmonary bypass, and extracorporeal membrane oxygenation.8,27-29
Regulatory Status
Numerous regulatory statutes and guidelines are concerned with DEHP levels in consumer products, allowable environmental levels, and limits of occupational exposure. In the Consumer Product and Safety Improvement Act of 2008, issued by Congress, and in a final ruling by the U.S. Consumer Product Safety Commission in 2017, any children’s toy or childcare articles are prohibited from containing concentrations of more than 0.1% of eight designated phthalates, including DEHP.30,31 FDA regulates the use of DEHP as an indirect food additive used in food-contact materials. DEHP can be used in semi-rigid and rigid acrylic plastic materials at levels up to 3% by weight.32 Additionally, DEHP can be a component of cellophane food packaging materials if DEHP levels alone or in combination with other phthalates do not exceed 5% by weight.33 EPA established a maximum contaminant level for DEHP in drinking water at 6 μg/L and an oral reference dose of 0.02 mg/kg/day on the basis of increased relative liver weights in exposed guinea pigs.34-36 Due to the potential for increased exposure via inhalation in occupational settings, the Occupational Safety and Health Administration (OSHA) has set an 8-hour time-weighted average permissible exposure limit of 5 mg/m3, which is equivalent to the limits recommended by the American Conference of Governmental Industrial Hygienists and the National Institute for Occupational Safety and Health.37 The short-term (15-minute) exposure limit allowable by OSHA is 10 mg/m3. The Agency for Toxic Substances and Disease Registry developed DEHP minimal risk levels of 0.1 and 0.06 mg/kg/day via an oral exposure route for intermediate and chronic exposure durations, respectively.4
Absorption, Distribution, Metabolism, and Excretion
Experimental Animals
Numerous studies have evaluated the absorption, distribution, metabolism, and excretion (ADME) properties of DEHP. High levels of hydrolase activity present in the intestinal tract of various mammalian species hydrolyze DEHP to its monoester form, MEHP, and 2-ethylhexanol. Endogenous hydrolytic activity has been shown to vary between species.38-41 In general, investigators believed that most of consumed DEHP is efficiently hydrolyzed to its monoester form prior to absorption in the intestinal tract, and that absorption of the diester form is associated with high-exposure levels that exceed the hydrolytic capacity of the intestinal pancreatic lipases. Albro et al.40 found no DEHP in the livers of rats after oral administration of DEHP at low doses (<0.4 g/kg), but did find detectable levels after administration of higher doses (>0.5 g/kg). Comparative studies in which male Sprague Dawley rats were administered DEHP by intraperitoneal injection (4 g/kg) or oral gavage (2 g/kg) revealed that approximately 80% of the oral dose undergoes mono-de-esterification compared to only 1% of the parenteral dose.42 Co-administration of a pancreatic lipase inhibitor (S,S,S-tributylphosphorothionate) resulted in a marked inhibition of DEHP intestinal absorption, suggesting MEHP is more readily absorbed than its parent molecule, DEHP.
In adult Wistar rats following a single oral administration of [14C]-DEHP (2.9 mg/kg), the dose was excreted primarily in the urine (42%) and feces (57%) by 7 days postadministration, with an estimated absorbed dose of 50% from the gastrointestinal tract.43 Dermal absorption efficiency of [14C]-DEHP is limited. Only an estimated 6.5% of a single 30–40 mg/kg dose in ethanol was absorbed by 7 days postapplication on exposed skin of male Fischer 344 (F344) rats.44 Numerous studies report little retention of radiolabeled DEHP or its metabolites in isolated tissues.38,45,46 However, elevated concentrations have been detected in rodent liver, adipose tissue, kidney, bladder, testis, and lungs; these findings may be associated with variables of study design such as the administered dose, route of exposure, or duration of exposure prior to necropsy.47-49 A comparative study in adult male Sprague Dawley rats, male dogs (beagles), and male miniature pigs (Hormel strain) reported differential DEHP excretion profiles following dietary exposure to 50 mg/kg/day for 21–28 days before administration of a single dose of [14C]-DEHP (50 mg/kg).46 Excretion of radioactivity in urine and feces by 24 hours postadministration was 27% and 57% (rats), 12% and 56% (dogs), and 37% and 0.1% (pigs), respectively; and after 4 days was 37% and 53% (rats), 21% and 75% (dogs), and 79% and 26% (mini-pigs), respectively. Overall elimination of radioactivity was complete by postadministration day 4 in all species and was most rapid in rats, followed by dogs, and least rapid in mini-pigs. Sjöberg et al.50 investigated the kinetics of DEHP and MEHP in Sprague Dawley rats following a single oral gavage of 1,000 mg/kg DEHP. In blood samples collected at 1, 3, 7, 9, 12, 15, 24, and 30 hours after dosing, DEHP was only detectable within the first 7 hours after dosing. The maximal plasma concentration (Cmax) of MEHP occurred within 1 hour of dosing (Cmax of 0.093 μg/mL), and a plasma elimination half-life of approximately 2.8–3.9 hours was determined. In another study, plasma Cmax of DEHP (8.8 μg/mL) and MEHP (63.2 μg/mL plasma) were reached within 6 hours of a single oral administration of DEHP in male Wistar rats (2.8 g/kg).51 In the same study, daily dosing for a week resulted in no accumulation of DEHP or MEHP in plasma.
Additional studies suggest that differential ADME properties during gestational and juvenile development could increase exposure in these sensitive subgroups. DEHP is able to cross the placental barrier,52,53 and maternal transfer of DEHP and its metabolites can occur via lactation.54 Increased intestinal tissue surface area relative to body weight and higher relative blood flow to the intestines may contribute to higher absorption rates in neonate/juvenile animals than in adults.55
Following hydrolysis of DEHP to MEHP by pancreatic lipases in the intestinal tract, MEHP can be further metabolized through oxidation to additional products and undergo subsequent conjugation with glucuronic acid. Interspecies variation in metabolic competencies can lead to distinct urinary metabolite profiles. Oral gavage of DEHP or MEHP to Sprague Dawley or F344 rats resulted in identification of over 20 distinct urinary metabolites.39,40 Phthalic diacids typically constitute most metabolites identified in rat urine.40 Rats differ from other tested species in that they display extensive oxidative metabolism of DEHP, but little capacity to conjugate these metabolites. In mice, exposure to MEHP resulted in detectable concentrations of MEHP and metabolite glucuronide conjugates in urine.56,57 Primates generally display reduced pancreatic lipase activity in the intestinal tract compared with rodents, leading to reduced conversion of DEHP to MEHP.58 Additionally, primates exhibit a reduced capacity to oxidize DEHP metabolites, but an increased capacity to conjugate (glucuronidate) MEHPmetabolites.58 Therefore, primates predominately excrete glucuronides of MEHP and metabolites with hydroxyl side chains that require limited oxidative metabolism.40
Humans
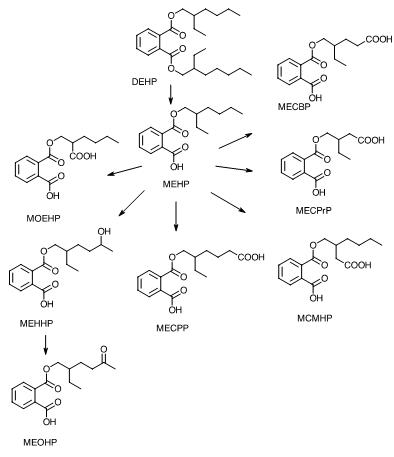
Studies have been conducted investigating DEHP toxicokinetic properties in humans. Similar to laboratory mammals, humans hydrolyze DEHP to MEHP by pancreatic lipases in the lumen of the intestinal tract, generate further oxidative metabolites, and conjugate these metabolites for excretion in urine and feces. In a study by Koch et al., urinary and serum concentrations of DEHP metabolites were determined from a human male volunteer following a single oral dose (0.64 mg/kg) of deuterium-labeled DEHP.59 Peak concentrations of three DEHP metabolites [MEHP, mono(2-ethyl-5-hydroxyhexyl) phthalate (MEHHP), and mono(2-ethyl-5-oxohexyl) phthalate (MEOHP)] were reached in serum after 2 hours and in urine after 4 hours. In serum, DEHP metabolites were unconjugated and contained high concentrations of MEHP relative to MEHP oxidation products. Urine samples contained higher concentrations of polar MEHP oxidation products than MEHP. Estimated serum elimination half-lives were <2 hours for the three measured DEHP metabolites. In a follow-up study, five DEHP urinary metabolites were identified [MEHP, MEHHP, MEOHP, mono(2-ethyl-5-carboxypentyl) phthalate (MECPP), and mono(2-[carboxymethyl]hexyl) phthalate (MCMHP)] that could be used as biomarkers for more accurate estimations of DEHP exposure (Figure 2).60 An additional three oxidative metabolites—mono(2-ethyl-3-carboxypropyl) phthalate (MECPrP), mono-2-(1-oxoethylhexyl) phthalate (MOEHP), and mono(2-ethyl-4-carboxybutyl) phthalate (MECBP)—were reported in human biomonitoring studies (Figure 2).61 Most of these metabolites undergo phase 2 metabolism to form glucuronide conjugates. Urinary concentrations of MEHHP and MEOHP have been detected at 10-fold higher concentrations than MEHP in 127 paired human samples, suggesting these metabolites may be more sensitive measures of DEHP exposure in the general population.62 Given these findings, DEHP exposure could be significantly underestimated in studies that measure only MEHP concentrations to predict human exposure. Other numerous oxidative metabolites of DEHP have also been proposed.61
Toxicity
Experimental Animals
Extensive literature exists on the toxicity of DEHP in numerous animal models. Acute oral median lethal dose (LD50) values for DEHP range from 9,800 to >40,000 mg/kg in rats,4,63-65 and 9,860 to >31,360 mg/kg in mice,4,63,66 LD50 values of 33,900 mg/kg in rabbits64 and 26,300 mg/kg in guinea pigs have also been reported.63,67 Neonatal and young animals may be more sensitive, however, to the acute effects of DEHP. Mortality was observed in 6- to 21-day-old male Sprague Dawley rats administered five daily oral doses of 1,000 or 2,000 mg DEHP/kg, whereas no mortality occurred in rats age 6 weeks or older when administered the same five daily doses.68
NTP has reported findings from studies investigating the acute, subchronic, and chronic toxicities of DEHP in rodent models.69 No effect on survival was observed in F344 rats or B6C3F1 mice during a 14-day observation period following a single administration of DEHP by oral gavage (800 to 20,000 mg/kg for rats; 1,252 mg/kg for mice). In 13-week feeding studies, F344 rats were administered a diet containing 0, 1,600, 3,100, 6,300, 12,500, or 25,000 ppm DEHP. Significantly reduced mean body weight gains were observed in male and female rats exposed to 25,000 ppm, and testicular atrophy was observed in males exposed to dietary concentrations of 12,500 ppm or higher. B6C3F1 mice exposed to 0, 800, 1,600, 3,100, 6,300, or 12,500 ppm DEHP in the diet for 13 weeks showed similar effects on body weight at the higher concentrations. Decreased mean body weight gains (≥10% relative to the control groups) were noted in male mice exposed to 3,100 ppm DEHP or higher and in all DEHP-exposed female mice, except the 1,600 ppm group. In a 2-year study, F344 rats were exposed to 0, 6,000, or 12,000 ppm DEHP in the diet, resulting in mean daily chemical intakes of 322 and 674 mg/kg body weight (mg/kg) for males, respectively; and 394 and 774 mg/kg for females, respectively. At the end of the 2-year study, mean body weights of exposed rats were up to 20% lower in the high-exposure groups compared to the control groups. In a companion 2-year study, B6C3F1 mice were exposed to 0, 3,000, or 6,000 ppm DEHP in the diet, resulting in mean daily chemical intakes of 672 and 1,325 mg/kg for males, respectively, and 799 and 1,821 mg/kg for females, respectively. At the end of the 2-year study, mean body weights were 7% and 10% lower in the 3,000 and 6,000 mg/kg male groups, and 21% and 33% lower in the 3,000 and 6,000 mg/kg female groups, respectively, relative to the control groups. The incidence of testicular tubule degeneration or atrophy was significantly elevated in high-exposure group male rats (approximately 90%) and male mice (approximately 14%) relative to the control groups.
Numerous laboratory animal studies have reported reductions in body weight and body weight gain following repeated exposures to DEHP, and common target organs of DEHP toxicity include the testis, kidney, and liver. Toxic effects of phthalates on the male reproductive tract are well characterized and are addressed in a subsequent section of this Introduction. DEHP effects on the kidney include reduced creatinine clearance, increased absolute and relative kidney weights, increased incidence and/or severity of mineralization of renal papilla, increased incidence and/or severity of tubule cell pigment, and increased incidence and/or severity of chronic progressive nephropathy.70-72 Liver enlargement due to both hepatocyte hyperplasia and hypertrophy, with associated morphological changes such as increased size and number of peroxisomes and corresponding increases in fatty-acid metabolism, are known hallmarks of DEHP toxicity in rodents. Activation of the peroxisome proliferator-activated receptor alpha (PPARα) in hepatocytes is recognized as a key molecular initiating event by which DEHP induces adverse effects in the liver.73 PPARα-deficient mice did not exhibit characteristic liver toxicity following 24 weeks of DEHP exposure but did exhibit moderate kidney and testicular toxicity.74 These findings suggest that while DEHP-induced liver toxicity is associated with PPARα status, renal and testicular toxicities likely manifest via alternative mechanisms.
Decreased severity of hepatic effects in nonrodent species may be related to interspecies differences in PPARα expression, binding, localization, and downstream molecular signaling pathways.41 DEHP metabolites such as MEHP have been reported to be more potent activators of human and mouse PPARα relative to its parent molecule.75 Therefore, interspecies differences in pancreatic lipase activity, which converts DEHP to MEHP, may influence observed DEHP toxicities. Additionally, routes of administration that bypass first pass metabolism in the intestinal tract and liver (intravenous), reducing hydrolysis of DEHP to its metabolites, could influence subsequent toxicity.
Humans
The health effects of DEHP have been evaluated extensively in animal models, but data that address the relationship between human health effects or adverse outcomes and exposure to DEHP are limited. Shaffer et al.64 presented a case report in which two male subjects had ingested single DEHP doses of 5 g and 10 g, respectively.64 The individual who consumed the 10 g dose presented with symptoms of mild gastric disturbance and moderate diarrhea, whereas no effects were observed at the lower dose.
Reproductive and Developmental Toxicity
Experimental Animals
Studies with laboratory rodents demonstrate that DEHP exposure can cause adverse effects on reproduction and development. In adult rats, oral DEHP exposure is associated with numerous deleterious effects on the male reproductive tract, including decreased weights of the testes, prostate, seminal vesicles, and epididymis; degeneration and atrophy of the seminiferous tubules; altered sperm parameters; and reduced fertility.4,13 The testes are considered a primary target tissue of DEHP toxicity. Decreased testicular weight and increased incidence of tubular atrophy have been observed in numerous rodent studies in which doses exceeded 100 mg/kg/day.69,76-82 Within the testes, DEHP appears to preferentially target Sertoli cell populations and directly or indirectly Leydig cell populations, which can impair spermatogenesis and fertility.78,83,84 Irregular seminiferous tubule structure and altered spermatogenesis were evident in male rats ingesting DEHP at 2,000 mg/kg/day via the diet for 15 days.85 In these rats, few spermatozoa were present in the lumen of the tubules, and damaged spermatogenic cells were observed in the tubular space. Significantly increased incidences of bilateral aspermatogenesis were observed at lower exposure concentrations (29 mg/kg/day) in male rats fed DEHP-supplemented diets for 2 years.86 Prepubertal rodents appear to be more sensitive to DEHP-mediated effects on the testes relative to sexually mature rodents.76,82 In contrast to studies in rodents, no changes in testes/epididymides weight or testicular histology were observed in cynomolgus monkeys following administration of 500 mg/kg/day DEHP by gavage for 14 consecutive days.87 Decreased fertility also has been observed in female rodents exposed to DEHP, and may be related to DEHP-induced disruption of normal estrous/ovulatory cycles.88
DEHP is a developmental toxicant in rodents, producing embryotoxic, fetotoxic, and teratogenic effects. Decreased fetal/pup body weight, increased rates of abortion and fetal resorptions, and malformations (hydronephrosis, cardiovascular malformations, and tail malformations) have been reported in rat dams and corresponding litters after exposure to DEHP during pregnancy/gestation.89,90 Exposure to DEHP during the perinatal period (gestation and/or lactation) can induce abnormal development of the male reproductive tract and other androgen-sensitive tissues. Although the exact mechanism is unknown, DEHP acts as an endocrine disruptor via an antiandrogenic mode of action and decreases insulin-like hormone 3 production by Leydig cells. Normally, during the window of fetal male sexual differentiation (gestation days 15.5–21.5), androgen-dependent masculinization of the fetal reproductive tract occurs, resulting in differentiation of the internal (epididymis, vas deferens, seminal vesicles, prostate) and external (penis, scrotum, perineum) genitalia.91,92 Exposure to DEHP during this critical window of susceptibility decreases fetal testosterone synthesis leading to structural malformations and functional alterations of the male reproductive system.93,94 Reduced anogenital distance (AGD), retained nipples, penile morphological abnormalities (hypospadias), undescended testes (cryptorchidism), small/absent sex accessory glands, delays in onset of pubertal landmarks (preputial separation), and histopathological alterations in testes and epididymides have been observed in male rats following perinatal DEHP exposure.93,95-97 Dysmorphogenic effects in the testes include microscopic disorganization of the seminiferous tubules with detachment of the spermatogonial cells from the basal membrane and absence of spermatocytes.98 The term “phthalate syndrome” is often used to describe the compendium of reproductive tract malformations observed in male test animals following in utero phthalate exposure.99
The reproductive and developmental effects of DEHP exposure were comprehensively investigated by NTP in a multigenerational reproductive assessment by continuous breeding study.100 In this study, DEHP was administered in the diet at concentrations of 1.5 (control), 10, 30, 100, 300, 1,000, 7,500, or 10,000 ppm to Sprague Dawley rats over multiple successive generations (F0, F1, F2, and F3) throughout the breeding, gestational, lactational, and postweaning intervals. Measured feed consumption and body weights informed the calculation of approximate daily doses of 0.12, 0.78, 2.4, 7.9, 23, 77, 592, and 775 mg/kg/day in the F0 animals; 0.09, 0.48, 1.4, 4.9, 14, 48, 391, and 543 mg/kg/day in the F1 animals; and 0.1, 0.47, 1.4, 4.8, 14, 46, and 359 mg/kg/day in the F2 animals. The 10,000 ppm group was removed from the study following the F1 generation due to the inability to produce offspring (F2 generation). Adverse reproductive and developmental effects such as decreased pregnancy index, decreased male AGD, delayed onset of pubertal landmarks (testes descent, vaginal opening, and balanopreputial separation), sperm counts, small male reproductive organs (testes, epididymis, and caudal epididymis), and seminiferous tubule atrophy were observed in all generations in the 7,500 and 10,000 ppm groups. No reproductive toxicity was evident at exposure concentrations <7,500 ppm; however, increased incidences of small testes and prostates were noted in 300 and 1,000 ppm male rats. After further review of animal studies by NTP’s Center for the Evaluation of Risks to Human Reproduction (CERHR) expert panel, a developmental no-observed-effect level of 1,000 ppm was suggested and calculated to be no more than 46 mg/kg/day based on the average dose over three generations.13
Humans
Given the results from animal studies, there is significant concern that DEHP can adversely affect human reproduction and male development. FDA’s CDRH concluded that “children undergoing certain medical procedures may represent a population at increased risk for the effects of DEHP.” A similar conclusion was reached by NTP’s CERHR, which found cause for “serious concern” that certain medical treatments may result in DEHP exposure levels that could adversely affect the development of the reproductive tract in male infants.13 Numerous epidemiological studies have found no significant association of DEHP or its metabolites with sperm abnormalities, circulating hormone concentrations, or indications of infertility.101-105 Other studies have reported associations between maternal urinary DEHP metabolite concentrations and effects on several markers of human male genital development. In complementary studies by Swan et al., measures of AGD and penile width in male infants were significantly associated with exposure to DEHP and three of its metabolites.106,107 Many parallels exist between the “phthalate syndrome” suite of effects in animal models and descriptions of human testicular dysgenesis syndrome. This syndrome is characterized by increased incidences of reproductive tract malformations in male newborns (cryptorchidism, hypospadias) and adverse effects in young adults (low sperm counts, testicular germ cell cancer) and is likely related to in utero exposure to environmental chemicals.108,109
Immunotoxicity
Experimental Animals
Several studies have been conducted to assess the potential of DEHP or MEHP to modulate immune function. Studies by Larsen et al. found subcutaneous injections of MEHP (100 μg) to induce an immunosuppressive effect characterized by a reduction in immunoglobulin E (IgE) and IgG1 antibody production in BALB/cJ mice following co-administration of ovalbumin antigen.110 However, in the same study, lower doses of MEHP (1 μg) induced an adjuvant effect characterized by increased IgE production. Administration of DEHP to male F344 rats via the diet (12 ppm) for 21 days resulted in a suppressed hepatic T-helper Type 1 (Th1) immune response initiated via intraperitoneal exposure to Mycobacterium bovis purified protein derivative.111 The authors hypothesized that this effect was associated with biotransformation of DEHP to MEHP and subsequent activation of PPARα. Further studies investigating the mixed T-helper cell adjuvant properties of DEHP found that this effect occurred independent of PPARα status in mouse models.112 Differential effects on the immune system have been noted in studies that use a developmental exposure paradigm. Increased sensitivity to DEHP exposure, characterized by altered immune parameters (T-dependent antigen response, natural killer cell activity, and tumor necrosis factor-alpha [TNF-α] production), was observed in male Wistar rats administered DEHP (0, 1, 3, 10, 30, 100, 300, or 1,000 mg/kg/day) by oral gavage during their juvenile period (postnatal day [PND] 10–50) relative to adult-only exposure (PND 50–90).113 In contrast, no persistent effects on numerous assessed immune parameters were noted in a study by Piepenbrink et al. in which CD rats were gestationally exposed to DEHP (0, 37.5, 75, 150, or 300 mg/kg/day).114 In the same study, no DEHP-associated immunotoxicity was noted in nulliparous exposed adults. Topical DEHP administration in adult B6C3F1 female mice did not increase serum concentrations of IgE, interleukin-4 (IL-4), or IL-13, suggesting a limited potential to induce allergic asthma.115 Additional studies report dose-dependent increases in some inflammatory cell numbers (macrophages, eosinophils, neutrophils, and/or lymphocytes) in bronchoalveolar lavage fluid in BALB/c or BALB/cJ mice following inhalation exposure to MEHP aerosols.116,117 Using median residential indoor air and worst-case exposure concentrations of 0.04 μg and 1.2 μg DEHP/m3, respectively, Hansen et al. estimated a margin of exposure between 2,500–75,000, suggesting that immune effects from inhalation exposures are only expected at air concentrations that are well in excess of environmental inhalation exposures typically encountered by humans.116
Humans
Numerous case reports and epidemiological studies suggest a link between phthalate exposure from PVC products and development of allergies and/or asthma.118 In a study of 39 PVC-processing plant workers, a higher prevalence of asthma, rhinitis, and eye and respiratory symptoms was observed in individuals exposed to PVC pyrolysis products and phthalates relative to an unexposed reference group.119 In a population-based incident case-control study of 521 new asthma cases and 932 control cases, asthma risk was related to the presence of plastic wall materials.120 Two epidemiological studies suggest childhood exposure to phthalates via house dust is related to the onset of allergy and/or asthma. In a nested case-control study within a 10,852 child cohort (198 persistent allergic cases, 202 control cases), higher median concentrations of DEHP (cases – 0.828 mg/g dust; control group – 0.723 mg/g dust) in house dust were significantly (p = 0.022) associated with asthma.121 In a separate study in Bulgarian preschool-age children (n = 102), wheezing was associated with higher DEHP concentrations in dust samples collected from children’s rooms (1.24 mg/g dust for children with wheezing, rhinitis, and/or eczema versus 0.86 mg/g dust for nonsymptomatic) in the preceding 12-month time interval.122 A greater understanding of human exposure relative to animal effect levels and additional mechanistic studies are needed to support a causal inference between DEHP exposure and immunomodulatory effects in humans.
Carcinogenicity
Experimental Animals
Multiple rodent studies were identified in the literature that examined the carcinogenic activity of DEHP, all of which initiated exposure once test animals had reached adulthood. Increased incidences of hepatocellular neoplasms have been corroborated across multiple rodent studies, along with reports of increased incidences of testicular Leydig cell tumors and pancreatic acinar adenomas in male rats following chronic exposure to DEHP. In 2-year cancer bioassays conducted by NTP, F344 rats and B6C3F1 mice were administered diets containing 0, 6,000, or 12,000 ppm DEHP and 0, 3,000, or 6,000 ppm DEHP, respectively.69,123 DEHP was found to be carcinogenic in both F344 rats and B6C3F1 mice on the basis of increased incidences of hepatocellular adenomas/carcinomas or neoplastic nodules in both males and females. Significantly increased incidences of hepatocellular adenoma or carcinoma (combined) in F344 rats and B6C3F1 mice were observed at lower DEHP exposure concentrations (male rats: 2,500 ppm; male mice: 500 ppm; female mice: 1,500 ppm) in chronic studies by David et al.86,124,125 Additionally, incidences of pancreatic acinar adenomas were increased in male F344 rats at the highest tested exposure concentration (12,500 ppm).86 A 159-week study in male Sprague Dawley (SD-CD) rats found that the high-exposure concentration of DEHP (6,000 ppm, or 300 mg/kg/day) increased the incidence of benign Leydig cell tumors.126
In rats, the combination of hepatocellular, pancreatic, and testicular tumors is often referred to as the “tumor triad” and is associated with sustained peroxisome proliferator activity.127 Although the definitive mode of action of DEHP-mediated carcinogenesis is undetermined, several key events, including activation of PPARα, perturbation of cellular proliferation and apoptosis, selective clonal expansion, and oxidative stress, are hypothesized to contribute to the onset of tumorigenesis.
Humans
The carcinogenic activity of DEHP in humans has been reviewed by numerous federal and international agencies. In the 14th Report on Carcinogens published by NTP, DEHP was listed as reasonably anticipated to be a human carcinogen based on sufficient evidence of carcinogenicity in experimental animals.128 EPA classified DEHP as a Group B2 carcinogen, probable human carcinogen, based on clear evidence of DEHP-mediated induction of liver tumors in rodent models.35 The International Agency for Research on Cancer (IARC) previously classified DEHP as “not classifiable as to its carcinogenicity to humans” (Group 3).73 The IARC determination was based on two assumptions: (1) that DEHP-induced hepatocellular cancer in rodents occurred as a result of induced peroxisome proliferation activity, and (2) that this mechanism is not relevant to humans due to lower PPARα expression and lack of observable peroxisome proliferation phenotypes in humans following exposure to known PPAR ligands. However, in light of new information about mechanisms of action, in 2011 IARC reclassified DEHP as a Group 2B carcinogen, indicating that there is “sufficient evidence of carcinogenicity in experimental animals” in combination with “no or limited epidemiological data.”129 The reevaluation included consideration of recent studies with novel transgenic mouse models, such as PPARα-null mice, humanized PPARα mice, and mice that express a constitutively active PPARα isoform in hepatocytes.130-132 These studies indicate that DEHP can induce hepatocarcinogenesis through a PPARα-independent mechanism, and that other molecular signaling pathways, not just activation of PPARα, likely contribute to the development of cancer.
Epidemiological studies that investigate a link between DEHP exposure and cancer endpoints are limited. In a case-control study of female breast cancer patients, increased cancer risk was associated with elevated urinary concentrations of the DEHP metabolite MECPP, but not other identified DEHP metabolites.133 Additional cancer epidemiology studies have been conducted in occupational groups where subjects had worked in PVC processing and plastic manufacturing facilities where increased exposure to phthalate plasticizers was probable.134-140 However, many of these studies lacked analytical assessment of exposure to specific phthalates, limiting the ability to determine a causal relationship between DEHP exposure and human cancer.
Genetic Toxicity
The genetic toxicity of DEHP has been extensively investigated and reviewed over many years (e.g., Huber et al., IARC, and Caldwell).141-143 Overall, DEHP shows limited evidence of genotoxic potential, and for the sporadic positive results that have been reported, associations are weak, not reproducible, obtained in a nonstandard test system, or qualified to some degree by the authors. MEHP, one of the main DEHP metabolites, does elicit positive responses, however, in some genotoxicity assays. An early study reported increases in revertant colonies in Salmonella typhimurium strain TA100 and Escherichia coli strain WP2 B/r treated with 2.5 and 5.0 mM MEHP, doses that induced marked cytotoxicity.144 More recently, MEHP was reported to generate reactive oxygen species and, consequently, DNA strand breaks in cultured AS52 cells145 as well as in cultured mouse Leydig tumor cells and in human prostate adenocarcinoma cells as measured by the comet assay.146,147 In both cell lines in the latter two studies, the parent compound DEHP (3 mM for 24 hours) was also reported to induce DNA damage, although the concentration tested was 1,000 times higher than the concentration tested of MEHP (3 uM). Similarly, DNA damage, measured by the comet assay, was also reported for DEHP in cultured HeLa cells treated with 96.6 µM DEHP for 24 hours.148
NTP has conducted several in vitro and in vivo genotoxicity assays with DEHP. Unpublished NTP data are included in Appendix D of this report. Published NTP studies, results of which are consistent with most published studies, showed no induction of gene mutations in any of several strains of S. typhimurium149,150 or in cultured mouse lymphoma L5178Y tk+/− cells.151 Additional bacterial mutation studies also reported negative results (e.g., Simon et al.152). Cytogenetic studies in cultured Chinese hamster ovary cells were negative for induction of chromosomal aberrations and were either positive or equivocal for induction of sister chromatid exchanges.153 In vitro chromosomal aberration studies, not conducted by NTP, in human leukocytes and human fetal lung cells with DEHP also showed no significant increases in chromosomally aberrant cells,154 as did chromosome aberration studies in Chinese hamster cells.155-157 Studies that assessed the ability of DEHP to induce sex-linked recessive lethal mutations in germ cells of male Drosophila melanogaster after exposure of either adults (via injection) or larvae (via feeding) were negative.158,159
Although sporadic reports of DNA damage or chromosomal effects following in vitro exposure to DEHP exist, results from in vivo studies are almost uniformly negative. In an in vivo comet assay conducted as part of the Japanese led multi-laboratory international validation effort for the assay, DEHP, administered by gavage at a top dose of 2,000 mg/kg/day for 3 days, did not induce DNA damage in cells obtained from the stomach, liver, and bone marrow of male Sprague Dawley rats.160 In addition, bone marrow samples from those rats showed no increase in the percentage of micronucleated erythrocytes, which are biomarkers of chromosomal damage. In another study, no increases in the frequencies of gpt and Spi(-) mutations were seen in DNA extracted from liver cells of gpt transgenic rats (both F344 and Sprague Dawley backgrounds) with exposure of up to 12,000 ppm DEHP exposure in the diet for 4 weeks, a concentration that produced generalized toxicity (e.g., increased liver weights).161 Similarly, an earlier study found that a 13-week Sprague Dawley gpt delta transgenic rats exposed to 12,000 ppm DEHP in the diet resulted in no increases in mutations in liver cell DNA.162
A study designed to investigate the potential for DEHP to induce unscheduled DNA synthesis (UDS) in liver cells of male B6C3F1 mice—a species that is sensitive to tumor induction by DEHP—found that exposures up to 500 mg/kg DEHP acutely or 6,000 ppm in the diet for up to 28 days did not induce UDS, measured using autoradiographic methods.163 The investigators also treated primary mouse hepatocytes in culture with up to 1.0 mM DEHP and observed no UDS at time points ranging from 12–48 hours posttreatment.
The negative results from the in vivo studies described above contrast with an earlier study by Singh et al. that reported a weak positive response in an in vivo rodent dominant lethal test using ICR mice. In that study, DEHP was administered via intraperitoneal injection at 66% of the acute LD50 dose, determined as 38.35 mL/kg.164 However, Jäckh et al.165 reported that a second rodent dominant lethal test that used similar doses of DEHP but administered the chemical via oral gavage showed no induction of dominant lethal mutations. They therefore suggested that the weak positive response in the Singh et al. study was likely related to nongenotoxic mechanisms, as covalent binding to DNA was not detected in liver cells of rats administered 14C- and 3H-labeled DEHP (500 mg/kg) by gavage.165
Study Rationale
In response to data gaps related to in utero and early life phthalate exposure and resultant adverse reproductive, developmental, and carcinogenic effects, NTP initiated a cancer hazard assessment for lifetime exposure to environmental phthalates. For DEHP, studies in rodents have established the gestational period as the time of greatest sensitivity to adverse developmental effects, specifically dysmorphogenesis of the male reproductive system. DEHP is a known rodent carcinogen; however, no previous cancer assessments have included exposure during the perinatal period (gestation and lactation). Therefore, it is unknown whether the carcinogenic response is altered when lifetime exposure encompasses these critical developmental windows.
NTP designed two 2-year studies in rats to evaluate whether DEHP lifetime exposure, including during the perinatal developmental period, would alter the dose response of the carcinogenic response relative to postweaning-only exposure. In these studies, DEHP was administered in dosed feed to mimic a common route of human exposure.
Materials and Methods
Procurement and Characterization of Di(2-ethylhexyl) Phthalate
Di(2-ethylhexyl) phthalate (DEHP) was obtained from Aldrich Chemical Company Inc. (St. Louis, MO) in a single lot (lot 01514TH) that was used in both 2-year studies. Identity, purity, and stability analyses were conducted by the analytical chemistry laboratory at RTI International (Research Triangle Park, NC) (Appendix A). Reports on analyses performed in support of the DEHP studies are on file at the National Institute of Environmental Health Sciences (NIEHS).
Lot 01514TH of the chemical, a clear liquid, was identified as DEHP by infrared (IR) spectroscopy, 1H and 13C nuclear magnetic resonance (NMR) spectroscopy, gas chromatography (GC) with mass spectrometry (MS) detection, and high-resolution MS (HRMS) (Table A-1). The IR spectrum was in good agreement with a reference spectrum and the structure was consistent with DEHP. Both 1H and 13C NMR spectra were consistent with reference and predicted spectra. The GC/MS spectra correlated well with the structure of DEHP and the HRMS resulted in measured mass within 0.5 ppm of the theoretical value. The elemental analysis was consistent with the composition of DEHP.
Karl Fisher titration determined the water content of lot 01514TH to be 0.145%. Ultra-performance liquid chromatography (UPLC) with photodiode array (PDA) detection and GC with flame ionization detection (FID) were used to determine a purity of 99.7% (Table A-1). The UPLC/PDA and GC/FID showed a minor peak accounting for 0.2% and 0.3%, respectively, of the total response in the chromatograms. Therefore, bulk purity was determined to be >99%.
Accelerated stability studies confirmed that lot 01514TH was stable for at least 2 weeks when stored in sealed glass vials at 5°C and 60°C. The bulk chemical of lot 01514TH was homogenized by shaking each of the 50 L plastic jugs for approximately 2 minutes and then transferred to 4 L amber glass storage bottles, which were stored at room temperature. Periodic reanalysis of the bulk chemical was performed during the studies by the study laboratory using high-performance liquid chromatography (HPLC) with ultraviolet (UV) detection, and no degradation was detected (Table A-1).
Preparation and Analysis of Dose Formulations
The dose formulations were prepared approximately monthly by mixing DEHP with NIH-07 or NTP-2000 feed (Table A-2). Both the perinatal and postweaning study (Study 1) and the postweaning-only study (Study 2) used dose formulations of 300, 1,000, 3,000, and 10,000 ppm. Formulations were stored in sealed plastic bag-lined containers at room temperature (approximately 25°C) for up to 42 days. The plastic bags used by the study laboratory in the preparation and storage of blank and dosed feed were determined to have no DEHP above the limit of detection of the assay (1.27 ppm).
Homogeneity studies of the dose formulations in a 72-kg NIH-07 feed batch (300 and 10,000 ppm) and in a 92-kg NTP-2000 feed batch (300, 3,000, and 10,000 ppm) were performed prior to animal studies by the study laboratory with HPLC/UV (Table A-1). Formulations were determined to be homogenous and stable for 42 days at room temperature and under simulated dosing conditions.
Periodic analysis of the DEHP dose formulations was conducted by the study laboratory using HPLC/UV to determine purity and concentration (Table A-3, Table A-4). All preadministration dose formulations were within 10% of the target concentrations. For the perinatal and postweaning study (Study 1), all postadministration dose formulations of DEHP were within 10% of target concentrations. In the postweaning-only study (Study 2), one sample collected from the residual feed in the feeder was below 10% of the target concentration (−12.3%). All other postadministration values were within 10% of the target concentrations.
Animal Source
Time-mated (F0) female Sprague Dawley (Hsd:Sprague Dawley SD) rats were obtained from Envigo (formerly Harlan Laboratories, Inc., Indianapolis, IN) for use in the perinatal and postweaning study (Study 1). Weanling (4 to 5 weeks old) male and female Sprague Dawley (Hsd:Sprague Dawley SD) rats were also obtained from Envigo for use in the postweaning-only study (Study 2).
Animal Welfare
Animal care and use are in accordance with the Public Health Service Policy on Humane Care and Use of Animals. All animal studies were conducted in an animal facility accredited by AAALAC International. Studies were approved by the Battelle (Columbus, OH) Animal Care and Use Committee and conducted in accordance with all relevant National Institutes of Health (NIH) and National Toxicology Program (NTP) animal care and use policies and applicable federal, state, and local regulations and guidelines.
Two-year Studies
Exposure Concentration Selection Rationale
Dietary exposure concentrations of 0, 300, 1,000, 3,000, or 10,000 ppm DEHP were selected based on previous data from an NTP multigenerational reproductive assessment by continuous breeding (RACB) study, which included a perinatal exposure paradigm in the Sprague Dawley rat model. In the RACB study, the highest tested exposure concentration (10,000 ppm) was well-tolerated by pregnant dams and did not affect litter size or pup survival to weaning. However, this exposure concentration induced significant numbers of reproductive tract and testicular malformations in the F1 male offspring and perturbed developmental androgen signaling as evidenced by decreased anogenital distance (AGD) and delayed attainment of puberty. In a previous NTP cancer bioassay using Fischer 344 (F344) rats, increased incidences of hepatocellular neoplasms occurred at exposure concentrations of 6,000 and 12,000 ppm DEHP. Together, these data suggest that the selected exposure concentrations are likely to induce a carcinogenic response and adequately challenge developmentally exposed test animals. To facilitate comparison of the results of the two 2-year studies, with and without perinatal exposure, both studies used the same exposure concentrations.
Perinatal and Postweaning Study in Rats (Study 1)
F0 female rats were 12 to 14 weeks old upon receipt. Evidence of mating is defined as gestation day (GD) 1; F0 females were received on GD 2 and held for 4 days. F0 females were randomly assigned to one of five exposure groups on GD 5 (n = 45/group). Randomization was stratified by body weight that produced similar group mean weights using PATH/TOX SYSTEM software (Xybion Medical Systems Co., Cedar Knolls, NJ).
F0 females were quarantined for 11 days after receipt. Ten nonmated females received with the time-mated females were designated for disease monitoring 11 days after arrival; samples were collected for serological analyses and the rats were euthanized, necropsied, and examined for the presence of disease or parasites. The health of the F1 rats was monitored during the study according to the protocols of the NTP Sentinel Animal Program (Appendix C). Pinworms (Syphacia spp.) were diagnosed in sentinel animals during routine health monitoring evaluations. Infected animals did not display clinical signs and no pathological lesions were noted in relation to the presence of the pinworms. Study animals did not receive medication for potential pinworm infection. Following this finding, NTP, in coordination with the testing laboratory, developed and implemented a successful plan of pinworm containment and eradication. NTP required the testing laboratories to actively monitor animals to ensure the continued exclusion of pinworms from all studies going forward. All other test results were negative.
Beginning on GD 6, F0 time-mated female rats were fed diets containing 0, 300, 1,000, 3,000, or 10,000 ppm DEHP throughout gestation and lactation. Groups of 50 F1 rats/sex/exposure concentration continued on in the study after weaning and were fed diets containing the same respective DEHP concentration for 2 years.
F0 female rats were housed individually during gestation and with their respective litters during lactation. Water and dosed feed were available ad libitum. F0 females were weighed on GDs 5, 6, 9, 12, 15, 18, and 21 and on lactation days (LDs) 1, 4, 7, 14, and 21. During gestation, feed consumption was continuously measured over 3-day intervals from GD 6 through GD 21 (GD 6–9, 9–12, 12–15, 15–18, and 18–21). The day of parturition was considered to be LD 0. On apparent GD 26, all time-mated female rats that failed to deliver were euthanized and the uteri were examined and stained for evidence of implantation. Total litter weight and litter weights by sex were collected on postnatal day (PND) 1. Individual F1 pups were weighed on PNDs 4, 7, 14, and 21. Clinical observations and survival were evaluated throughout lactation. During lactation, feed consumption was continuously measured over 3-day intervals from LD 1 through LD 21 (LD 1–4, 4–7, 7–10, 10–14, 14–17, 17–21).
Select dams and their litters were removed on GD 18 to quantify mono(2-ethylhexyl) phthalate plasma and tissue concentrations. On GD 18, blood was collected from the retroorbital sinus of randomly selected dams (n = 5 per exposure group). Blood samples were collected in tubes containing K3 EDTA (tripotassium ethylene diamine tetraacetic acid), centrifuged, and the plasma harvested. Amniotic fluid was collected and pooled by dam, and each dam’s fetuses were collected and pooled by litter. All samples were flash frozen in liquid nitrogen and stored frozen at approximately −20°C before shipment to RTI International (Research Triangle Park, NC). All samples were analyzed using a validated analytical method (Appendix E).
On PND 4, all litters with surviving pups were retained. Before weaning, two males and two females per litter from 25 litters in the 0, 300, 1,000, and 3,000 ppm groups and from 21 litters in the 10,000 ppm group were randomly assigned to continue on in the 2-year postweaning phase of the study. To complete assignment in the 10,000 ppm group, two male pups and three female pups were selected from two litters, and two male pups and one female pup were selected from two additional litters. After assignments to the 2-year study were complete, 20 pups per sex from the remaining control pups were randomly selected as the sentinel animals. On the day the last litter reached PND 18, litters were randomly selected and F1 pups from these litters were randomly selected for the 2-year study. On the day the last litter reached PND 21, dams were removed and the pups were weaned. Weaning marked the beginning of the 2-year chronic phase of the study.
After weaning, F1 pups were housed up to two (males) or four (females) per cage. Dosed feed and water were available ad libitum. Feed consumption was measured weekly for the first 13 weeks and at 4-week intervals thereafter. Cages were changed weekly through PND 4, then changed at least twice weekly. Racks were changed and rotated at least every 2 weeks. Further details of animal maintenance are given in Table 1.
Two diets were utilized in this study: (1) NIH-07 during the perinatal phase, and (2) NTP-2000 during the postweaning phase. The NIH-07 diet is a higher protein diet that supports reproduction and lactation in rodents, whereas the NTP-2000 diet is a lower protein diet that decreases the incidence of chronic nephropathy in adult rats. Information on feed composition and contaminants for both diets is provided in Appendix B.
Postweaning-only Study in Rats (Study 2)
Male and female rats were 4 to 5 weeks old upon receipt and quarantined for 13 days prior to study start. Rats were randomly assigned to one of five exposure groups (n = 50 rats/sex/exposure group). Randomization was stratified by body weight that produced similar group mean weights using PATH/TOX SYSTEM software (Xybion Medical Systems Corporation, Cedar Knolls, NJ). Rats were 6 to 7 weeks old on the first day of the study and were provided DEHP in dosed feed for 2 years at one of five exposure concentrations (0, 300, 1,000, 3,000, or 10,000 ppm).
Five male and five female rats were designated for disease monitoring 13 days after arrival; samples were collected for serological analyses, and the rats were euthanized, necropsied, and examined for the presence of disease or parasites. The health of the rats was monitored during the study according to the protocols of the NTP Sentinel Animal Program (Appendix C). Pinworms (Syphacia spp.) were diagnosed in sentinel animals during routine health monitoring evaluations. All other test results were negative.
Rats were housed up to two (males) or four (females) per cage. Water and dosed feed were available ad libitum. Feed consumption was measured weekly for the first 13 weeks and at 4-week intervals thereafter. Cages were changed at least twice weekly. Racks were changed and rotated at least every 2 weeks. Further details of animal maintenance are given in Table 1. Information on feed composition and contaminants is given in Appendix B.
Clinical Examinations and Pathology
In both of the 2-year studies, animals were observed twice daily for morbidity and moribundity. Animals were weighed initially, weekly for the next 13 weeks, every 4 weeks thereafter, and at study termination. Beginning on study day 36 (Study 1) or study week 5 (Study 2), clinical observations were recorded every 4 weeks and at the end of the studies.
Complete necropsies and microscopic examinations were performed on all F1 rats in Study 1 and all rats in Study 2. At necropsy, all organs and tissues were examined for grossly visible lesions, and all major tissues were fixed and preserved in 10% neutral buffered formalin except for eyes, testes, vaginal tunics, and epididymides, which were first fixed in Davidson’s solution or modified Davidson’s solution. Tissues were processed and trimmed, embedded in paraffin, sectioned at a thickness of 4 to 6 μm, and stained with hematoxylin and eosin (H&E) for microscopic examination. For all paired organs (e.g., adrenal gland, kidney, ovary), samples from each organ were examined. In the original evaluation of the uterus, a transverse section through each uterine horn, approximately 0.5 cm cranial to cervix, was collected for histopathology evaluation. For the residual tissue evaluation of the uterus, all remaining uterine, including the cervix, and vaginal tissue was sectioned longitudinally and examined histologically. Results from the residual uterine evaluation were combined with those from the original, transverse section of uterus. Tissues examined microscopically are listed in Table 1.
Microscopic evaluations were completed by the study laboratory pathologist, and the pathology data were entered into the Toxicology Data Management System. The report, slides, paraffin blocks, residual wet tissues, and pathology data were sent to the NTP Archives for inventory, slide/block match, wet tissue audit, and storage. The slides, individual animal data records, and pathology tables were evaluated by quality assessment (QA) pathologists at a pathology laboratory independent of the study laboratory. The individual animal records and tables were compared for accuracy, the slide and tissue counts were verified, and the histotechnique was evaluated. For both 2-year studies, the QA pathologists evaluated slides from all neoplasms and all potential target organs, which included the liver, pancreas, kidney, heart, bone marrow, and pituitary gland of rats; testes and epididymis of male rats; and the uterus of female rats. Kidney pathology is reported only for Study 1. Additional sex-specific target tissues identified in Study 1 included the prostate glands, gubernacula, phallus, prepuce, seminal vesicles, and vagina.
The QA report and the reviewed slides were submitted to the NTP Pathology Working Group (PWG) coordinator, who reviewed the selected tissues and addressed any inconsistencies in the diagnoses made by the laboratory and QA pathologists. Representative histopathology slides containing examples of lesions related to chemical administration, examples of disagreements in diagnoses between the laboratory and QA pathologists, or lesions of general interest were presented by the QA/PWG coordinators to the PWG for review. The PWG consisted of the QA pathologists and other pathologists experienced in rodent toxicological pathology. The PWG examined the tissues without any knowledge of exposure groups. When the PWG consensus diagnosis differed from that of the laboratory pathologist, the diagnosis was changed. Final diagnoses for reviewed lesions represent a consensus between the laboratory pathologist, reviewing pathologist(s), and the PWG. Details of these review procedures have been described, in part, by Maronpot and Boorman166 and Boorman et al.167 For subsequent analyses of the pathology data, the decision whether or not to evaluate the diagnosed lesions for each tissue type separately or combined was generally based on the guidelines of McConnell et al.168
Benchmark Dose Analysis
Benchmark doses (BMDs) were calculated using the EPA Benchmark Dose Software (BMDS), version 3.1.2.169 and are presented in Appendix F. The dose variable for the models was the amount of DEHP consumed in mg/kg body weight/day (mg/kg/day). Numbers of animals per exposure group were poly-3-adjusted survival numbers. The response variable was the incidence of the endpoint being modeled.
All of the frequentist dichotomous models in the BMDS were used. The logistic, log-probit, and probit models were used with no parameter restrictions. Other models (dichotomous Hill, gamma, log-logistic, multistage, and Weibull) were used with default restrictions on the ranges of some of the parameters, as described in the BMDS User Guide.170
The benchmark response (BMR) used in the models was 0.1 (10%) extra risk, with estimated background levels. The benchmark dose lower confidence limit (BMDL) was calculated using a 95% confidence interval. The decision logic used to recommend one model from the fitted models was the default logic.170
Statistical Methods
Survival Analyses
The probability of survival was estimated by the product-limit procedure of Kaplan and Meier171 and is presented graphically. Animals surviving to the end of the observation period are treated as censored observations, as are animals dying from unnatural causes within the observation period. Animals dying from natural causes are included in analyses and are treated as uncensored observations. For the postweaning-only study (Study 2), exposure concentration-related trends are identified with Tarone’s life-table test,172 and pairwise exposure concentration-related effects are assessed using Cox’s method.173 For the perinatal and postweaning study (Study 1), exposure concentration-related trends and pairwise exposure-related effects on survival are assessed using a Cox proportional hazards model173 with a random litter effect. All reported p values for the survival analyses are two-sided.
Calculation of Incidence
The incidences of neoplasms or nonneoplastic lesions are presented as the numbers of animals bearing such lesions at a specific anatomic site. For calculation of incidence rates, the denominator for most neoplasms and all nonneoplastic lesions is the number of animals where the site was examined microscopically. When macroscopic examination was required to detect neoplasms in certain tissues (e.g., mesentery, pleura, peripheral nerve, skeletal muscle, tongue, tooth, and Zymbal’s gland) before microscopic evaluation, however, the denominator consists of the number of animals that had a gross abnormality. When neoplasms had multiple potential sites of occurrence (e.g., leukemia or lymphoma), the denominator consists of the number of animals on which a necropsy was performed. Additional study data also give the survival-adjusted neoplasm rate for each group and each site-specific neoplasm. This survival-adjusted rate (based on the Poly-3 method described below) accounts for differential mortality by assigning a reduced risk of neoplasm, proportional to the third power of the fraction of time on study, only to site-specific, lesion-free animals that do not reach terminal euthanasia.
Analysis of Neoplasm and Nonneoplastic Lesion Incidence
Statistical analyses of neoplasm and nonneoplastic lesion incidence considered two features of the data. Some animals did not survive the entire 2 years of the study, so survival differences between groups had to be considered. Also, up to two animals per sex were randomly selected from each litter to participate in the study, except for the 10,000 ppm group in the perinatal and postweaning study (Study 1) for which additional males and females were needed to populate the study. The statistical analysis of lesion incidence used the Poly-3 test to account for survival differences, with a Rao-Scott adjustment for litter effects, as described below.
The Poly-k test174-176 was used to assess neoplasm and nonneoplastic lesion prevalence. This test is a survival-adjusted quantal-response procedure that modifies the Cochran-Armitage linear trend test to account for survival differences. More specifically, this method modifies the denominator in the quantal estimate of lesion incidence to approximate more closely the total number of animal years at risk. For analysis of a given site, each animal is assigned a risk weight. This value is 1 if the animal had a lesion at that site or if it survived until terminal euthanasia; if the animal died before terminal euthanasia and did not have a lesion at that site, its risk weight is the fraction of the entire study time that it survived, raised to the kth power.
This method yields a lesion prevalence rate that depends only on the choice of a shape parameter for a Weibull hazard function describing cumulative lesion incidence over time.174 Unless otherwise specified, a value of k = 3 was used in the analysis of site-specific lesions. This value was recommended by Bailer and Portier174 after an evaluation of neoplasm onset time distributions for a variety of site-specific neoplasms in control F344 rats and B6C3F1 mice.177 Bailer and Portier174 showed that the Poly-3 test provided valid results if the true value of k is anywhere in the range from 1 to 5. A further advantage of the Poly-3 method is that it does not require lesion lethality assumptions. Variation introduced by the use of risk weights, which reflect differential mortality, was accommodated by adjusting the variance of the Poly-3 statistic as recommended by Bieler and Williams.178 Poly-3 tests used the continuity correction described by Nam.179
Littermates tend to be more like each other than like fetuses/pups in other litters. Failure to account for correlation within litters leads to underestimates of variance in statistical tests, resulting in higher probabilities of Type I errors (“false positives”). Because up to two pups per sex per litter were present in the perinatal and postweaning study (Study 1), the Poly-3 test was modified to accommodate litter effects using the Rao-Scott approach.180 The Rao-Scott approach accounts for litter effects by estimating the ratio of the variance in the presence of litter effects to the variance in the absence of litter effects. This ratio is then used to adjust the sample size downward to yield the estimated variance in the presence of litter effects. The Rao-Scott approach was implemented in the Poly-3 test as recommended by Fung et al.,181 formula ₸RS2.
Tests of significance included pairwise comparisons of each exposed group with control groups and a test for an overall exposure concentration-related trend. Continuity-corrected Rao-Scott-adjusted Poly-3 tests were used in the analysis of lesion incidence and reported p values are one-sided. The significance of a lower incidence or negative trend in lesions is approximated as 1 − p with the letter N added (e.g., p = 0.99 is presented as p = 0.01N). For neoplasms and nonneoplastic lesions observed without litter structure (e.g., at the interim evaluation), Poly-3 tests that included the continuity correction, but without adjustment for potential litter effects, were used for trend and pairwise comparisons to the control group.
To evaluate incidence rates by litter, the proportions of litters affected by each lesion type were tested among groups. The Cochran-Armitage trend test and Fisher’s exact test182 were used to test for trends and pairwise differences from the control group, respectively.
Analysis of Continuous Variables
Before statistical analysis, extreme values identified by the outlier test of Dixon and Massey,183 for small samples (n < 20), and Tukey’s outer fences method,184 for large samples (n ≥ 20), were examined by NTP personnel, and implausible values were eliminated from the analysis. Organ and body weight measurements, which historically have approximately normal distributions, were analyzed with the parametric multiple comparison procedures of Dunnett185 and Williams.186,187 Dam gestational and lactational feed consumption, litter sizes, pup survival, implantations, number of resorptions, and proportions of male pups per litter for all studies were analyzed using the nonparametric multiple comparison methods of Shirley188 [as modified by Williams189] and Dunn190 given that these endpoints typically have skewed distributions. For all quantitative endpoints unaffected by litter structure, the Jonckheere test191 was used to assess the significance of exposure concentration-related trends and to determine, at the 0.01 level of significance, whether a trend-sensitive test (the Williams or Shirley test) was more appropriate for pairwise comparisons than a test that does not assume a monotonic exposure concentration-related trend (the Dunnett or Dunn test).
Postweaning body weights were measured on two pups/sex/litter in most cases in the perinatal and postweaning study (Study 1); more than two pups/sex/litter were common in preweaning body weight measurements. The analyses of pup mean body weights and mean body weights adjusted for litter size (described below) of these animals took litter effects into account using a mixed model with litter as a random effect. To adjust for multiple comparisons, a Dunnett-Hsu adjustment was used.192 Dam mean body weights during gestation and lactation were analyzed with the parametric multiple comparison procedures of Dunnett185 and Williams,186,187 depending on whether the Jonckheere test indicated the use of a trend-sensitive test. P values for these analyses are two-sided.
Analysis of Gestational and Fertility Indices
Cochran-Armitage trend tests were used to test the significance of trends in gestational and fertility indices across exposure groups. Fisher’s exact test was used to conduct pairwise comparisons of each exposed group with the control group. P values for these analyses are two-sided.
Body Weight Adjustments
To adjust preweaning pup body weights for live litter size, a linear model was fit to body weights as a function of exposure concentration and litter size. The estimated coefficient of litter size was then used to adjust each pup body weight on the basis of the difference between its litter size and the mean litter size. Preweaning pup body weights were adjusted for PND 1 live litter size. After adjustment, mean body weights were analyzed with a linear mixed model with a random litter effect.
Historical Control Data
The concurrent control group is the most valid comparison to the exposed groups and is the only control group analyzed statistically in NTP bioassays. However, historical control data are often helpful in interpreting potential exposure-related effects, particularly for uncommon or rare neoplasm types. For meaningful comparisons, the conditions for studies in the historical control data must be generally similar. Significant factors that can affect the background incidence of neoplasms at a variety of sites are diet, sex, strain/stock, and route of exposure. The NTP historical control database contains all 2-year studies for each species, sex, and strain/stock with histopathology findings in control animals completed within the most recent 5-year period,193-195 including the concurrent control for comparison across multiple technical reports. In general, the historical control data for a given study includes studies using the same route of administration, and the overall incidence of neoplasms in control animals for all routes of administration are included for comparison, including the current study.
Quality Assurance Methods
Both the perinatal and postweaning study (Study 1) and the postweaning-only study (Study 2) were conducted in compliance with the Food and Drug Administration Good Laboratory Practice Regulations.196 In addition, both study reports were audited retrospectively by an independent QA contractor against study records submitted to the NTP Archives. Separate audits covered completeness and accuracy of the pathology data, pathology specimens, final pathology tables, and a draft of this NTP Technical Report. Audit procedures and findings are presented in the reports and are on file at NIEHS. The audit findings were reviewed and assessed by NTP staff, and all comments were resolved or otherwise addressed during the preparation of this Technical Report.
Genetic Toxicology
A large number of genetic toxicity tests for DEHP were conducted by NTP. The genetic toxicity data presented here are the culmination of several NTP evaluations of DEHP that have not been previously published. The protocols used for the conduct and evaluation of the in vivo chromosomal aberrations and micronucleus tests are described in detail in Appendix D.
The genetic toxicity studies have evolved from an earlier effort by NTP to develop a comprehensive database permitting a critical anticipation of a chemical’s carcinogenicity in experimental animals based on numerous considerations, including the molecular structure of the chemical and its observed effects in short-term in vitro and in vivo genetic toxicity tests (structure-activity relationships). The short-term tests were originally developed to clarify proposed mechanisms of chemical-induced DNA damage on the basis of the relationship between electrophilicity and mutagenicity197 and the somatic mutation theory of cancer.198,199 It should be noted, however, that not all cancers arise through genotoxic mechanisms.
DNA reactivity combined with Salmonella mutagenicity is highly correlated with induction of carcinogenicity in multiple species/sexes of rodents and at multiple tissue sites.200 Information from other in vitro genotoxicity assays does not appear to increase the predictivity of the bacterial mutation assay for rodent carcinogenicity, but these other tests can provide useful information on the types of DNA and chromosomal damage induced by the chemical under investigation. Positive results seen in in vivo assays that measure induction of chromosomal damage have been shown to have a high correlation with rodent carcinogenicity.201
Results
Data Availability
The National Toxicology Program (NTP) evaluated all study data. Data relevant for evaluating toxicological findings are presented here. All study data are available in the NTP Chemical Effects in Biological Systems (CEBS) database: https://doi.org/10.22427/NTP-DATA-TR-601.202
Perinatal and Postweaning Study in Rats (Study 1)
Perinatal Phase
No effects on maternal survival were observed following exposure to di(2-ethylhexyl) phthalate (DEHP), and no exposure-related maternal clinical observations were noted (Appendix H). Administration of DEHP had no effects on the percentage of pregnant females that produced pups, gestation length, or pup sex distribution (Table 2; Appendix H). The lower number of females that produced pups in the 10,000 ppm group was due to 13 mated females that were not pregnant. This was not attributed to the test article, because dam exposure to DEHP started after the period of implantation.
Mean gestation body weights of dams receiving up to 3,000 ppm DEHP in the diet were within approximately 3.5% of control animals (Table 3). Dams that received 10,000 ppm DEHP in the diet displayed significantly decreased mean body weights (up to 10%), relative to control animals, throughout the gestational period. Lower relative body weights in 10,000 ppm dams were associated with significantly decreased body weight gain over the GD 6–9, GD 15–18, and GD 18–21 intervals (Table 3). During the gestational period (GD 6–21), the overall mean body weight gain of 10,000 ppm dams was significantly decreased 27% compared to that of the control animals. During the lactational period, there were no effects on maternal mean body weight or body weight gain among dam groups receiving up to 3,000 ppm DEHP (Table 3). Lactational body weights of 10,000 ppm dams were significantly decreased (up to approximately 25%) at all assessed lactational time points relative to control animals. This decrease was more severe in magnitude than what was observed during gestation and is likely the result of decreases in absolute body weight during the lactational period. All other exposure groups, including the control group, displayed positive weight gains during the lactation day (LD) 4–21 interval, whereas the 10,000 ppm dams lost an average of 24 g in body weight, corresponding to an approximate 10% decrease in body weight from LD 1 to LD 21.
For dams exposed at 10,000 ppm DEHP, significantly decreased feed consumption during both gestation (GD 6–21; approximately 14%) and lactation (LD 1–14; 39%), relative to control animals (Table 4), likely contributed to the observed decrements in body weight (Table 3). Decreased feed consumption (8%) was also observed in 3,000 ppm dams during the LD 17–21 interval, and attained statistical significance, relative to control animals. Gestational DEHP intake (GD 6–21) for dams in the 300, 1,000, 3,000, and 10,000 ppm groups was approximately 21, 68, 206, and 626 mg DEHP/kg body weight/day (mg/kg/day), respectively (Table 4). Lactational DEHP intake (LD 1–14) for dams in the 300, 1,000, 3,000, and 10,000 ppm groups was approximately 49, 166, 482, and 1,244 mg/kg/day, respectively (Table 4). Chemical intake for the LD 14–21 interval was not calculated due to the unknown contribution of offspring feed consumption to the overall cage-based measurements.
On postnatal day (PND) 1, total litter size and total live litter size of the 10,000 ppm group were significantly decreased, relative to the control group, corresponding to a reduction of approximately two pups per litter (Table 5). This litter effect corresponded to a significantly decreased number of live female offspring in 10,000 ppm DEHP-exposed litters. A significant decrease in the survival ratio (PND 5–21) of offspring exposed to 3,000 ppm was observed; however, this effect was not considered related to exposure because the survival ratio at 10,000 ppm was not different from that of the control group. No other effect of DEHP exposure on offspring survival was observed during the preweaning intervals (PND 1–4, PND 5–21, or PND 1–21) (Table 5).
Decreased PND 1 pup mean body weights were observed in male, female, and combined (male + female) offspring in 3,000 and 10,000 ppm DEHP litters (Table 6). PND 1 pup mean body weights (male, female, and combined) were significantly decreased by approximately 4% in the 3,000 ppm group, and by 15%, 12%, and 13% in males, females, and combined offspring in the 10,000 ppm group, respectively, relative to control animals. At weaning (PND 21), male and female pup mean body weights in the 1,000 and 3,000 ppm DEHP groups were significantly decreased approximately 6% compared to control animals. Severe effects on growth were evident in male and female offspring exposed to 10,000 ppm DEHP. At weaning (PND 21), mean body weights of male and female pups in the 10,000 ppm group were significantly decreased approximately 55% and 53% relative to those of control animals, respectively (Table 6). Significantly decreased mean body weights in 10,000 ppm pups were attributed to reduced body weight gain throughout the preweaning interval. Pup survival was not reduced, and there were no exposure-related clinical observations. Offspring from the 10,000 ppm group were therefore continued on study.
Concentrations of the DEHP metabolite mono(2-ethylhexyl) phthalate (MEHP) were determined in maternal plasma, amniotic fluid, and fetal tissues collected on GD 18 (Table 7) using validated analytical methods (Appendix E). MEHP concentrations increased proportionally to exposure concentration in dam plasma in groups exposed to lower dietary DEHP concentrations (300 and 1,000 ppm). However, at higher dietary concentrations (3,000 and 10,000 ppm), the increase was greater than proportional to exposure concentration, despite proportional increases in chemical consumption, suggesting potential saturation of clearance pathways of MEHP (Figure 2). MEHP was measured in amniotic fluid and fetuses demonstrating transfer of MEHP across the placental barrier and exposure of the developing conceptus. The concentrations in fetuses were approximately 18%–28% of the plasma concentrations in dams, suggesting gestational transfer was moderate for MEHP, but the rate of transfer of other metabolites was not measured. MEHP was detected in amniotic fluid and fetus samples from control animals, whereas control dam plasma concentrations were below the limit of detection.
Postweaning Phase
Overall, survival at study termination of males and females in exposed groups was commensurate with control groups (Table 8; Figure 3). However, 6 to 7 rats per sex in the 10,000 ppm group died postweaning during the first two weeks of the study period. All but one of these early losses likely resulted from the severely reduced body weights of those animals.
At study termination, group mean body weights for male and female rats in the 300, 1,000, and 3,000 ppm DEHP groups were within 10% of their respective control groups (Table 9, Table 10; Figure 4). During the first 3 (females) or 6 (males) weeks on study postweaning, mean body weights of rats in the 10,000 ppm groups were approximately half that of their respective control groups. Following week 3 (females) or week 6 (males) on study, however, mean body weights of those male and female rats exhibited a moderate recovery, attaining maximum group mean body weights that were 28% and 17% lower, respectively, relative to control animals during the chronic study period. The terminal mean body weights of 10,000 ppm males and females were 30% and 32% lower than those of control animals, respectively. Lower body weights observed in the 10,000 ppm groups at study termination were attributed to reduced body weight gain observed over the duration of the study.
Feed consumption by male and female rats in the 300, 1,000, and 3,000 ppm DEHP groups was commensurate to that of the control groups throughout the study (Table 11, Table 12; Appendix H). Feed consumption was generally lower in the 10,000 ppm male and female rat groups with the largest difference directly following weaning. This finding was restricted to the early time interval and likely resulted from the significantly reduced body size of animals in the highest exposure group. Dietary concentrations of 300, 1,000, 3,000, and 10,000 ppm resulted in average daily doses of approximately 18, 58, 189, and 678 mg/kg/day for males and 18, 62, 196, and 772 mg/kg/day for females (Appendix H).
No exposure-related clinical findings were observed in any of the exposed groups (Appendix H).
Pathology
This section describes statistically significant or biologically noteworthy changes in the incidences of gross lesions, neoplasms, and/or nonneoplastic lesions of the liver, pancreas, male and female reproductive organs, kidney, heart, bone marrow, and pituitary gland. Summaries of the incidences of neoplasms and nonneoplastic lesions, individual animal neoplasm diagnoses, and statistical analyses of primary neoplasms that occurred with an incidence of at least 5% in at least one animal group are presented as supplemental data in Appendix H.
Liver: There were significant increases in the incidences of hepatocellular adenoma (10,000 ppm males and 3,000 ppm females) and carcinoma (10,000 ppm females) relative to control animals and a positive trend in incidence with increasing exposure concentration in males for hepatocellular carcinomas (Table 13). The incidences of hepatocellular adenoma or carcinoma (combined) were significantly increased in the 10,000 ppm male and female groups and in the 3,000 ppm female group relative to control animals. Hepatocellular adenomas were characterized by regions that were sharply demarcated from surrounding liver parenchyma, nodular, and compressing adjacent normal hepatocytes, with loss of normal lobular architecture and an irregular growth pattern. The liver plates typically impinged obliquely on the surrounding liver parenchyma. The hepatocytes within an adenoma generally varied in size. Hepatocellular carcinomas were characterized by one or more of the following features: local infiltrating growth and/or distinct lack of demarcation with the adjacent tissue, the presence of trabeculae composed of multiple layers of hepatocytes, cellular pleomorphism, loss of normal lobular architecture, regions of hemorrhage and/or necrosis, and increased mitotic figures.
There were significant increases in the incidences of many nonneoplastic liver lesions in DEHP-exposed groups relative to the control groups (Table 13). The incidences of hepatocellular cytoplasmic alteration were significantly increased in 3,000 and 10,000 ppm males and in all exposed groups of females relative to control animals. There were significant increases in the incidence of hepatocellular hypertrophy in the 10,000 ppm males and in the 1,000, 3,000, and 10,000 ppm females. Significant increases in the incidence of pigment were observed in the 1,000, 3,000, and 10,000 ppm males and in all exposed groups of females. There were significant increases in the incidence of hepatocellular necrosis in the 10,000 ppm males relative to control animals. The incidence of hepatocellular eosinophilic foci was significantly increased in 10,000 ppm females over that of the control group, and a positive trend was seen in males with increasing exposure concentration. The incidence of hepatocellular basophilic foci was significantly increased in the 10,000 ppm males relative to the control group, and a positive trend with increasing exposure occurred in females. There was a significant increase in the incidence of bile duct hyperplasia in 3,000 ppm females compared to control animals.
Hepatocellular cytoplasmic alteration was characterized by hepatocytes that were expanded with eosinophilic granular cytoplasm (Figure 5). A four-grade severity scale reflected the degree of tissue affected in the section of liver that was evaluated histologically: minimal (grade 1), up to 25% of hepatocyte involvement; mild (grade 2), 26% to 50% of hepatocyte involvement; moderate (grade 3), 51% to 75% of hepatocyte involvement; and marked (grade 4), at least 76% of hepatocyte involvement. Hepatocellular hypertrophy often occurred in conjunction with cytoplasmic alteration and/or pigment. Hypertrophy was characterized by enlargement of the hepatocytes. In lesser affected animals, hypertrophy was confined to centrilobular regions, but in more severely affected animals, hypertrophy extended into the midzonal and periportal areas. A four-grade severity scale was used: minimal (grade 1), up to 10% of hepatocyte involvement; mild (grade 2), 11% to 25% of hepatocyte involvement; moderate (grade 3), up to 50% of hepatocyte involvement; and severe (grade 4), >51% of hepatocyte involvement. Hypertrophy was generally centrilobular and often involved only a few cells per lobule. Although hypertrophy was only occasionally observed in males (at the 3,000 and 10,000 ppm exposure concentrations), in females it exhibited a concentration-responsive significant increase in incidence (but not severity) starting at the 1,000 ppm exposure concentration.
Pigment only occurred in exposed animals and was characterized by a pale gold-colored pigment within the hepatocellular cytoplasm (Figure 5). A four-grade severity scale was used: minimal (grade 1), up to 30% of hepatocytes contained pigment; mild (grade 2), 31% to 50% of hepatocytes contained pigment; moderate (grade 3), >51% of hepatocytes contained pigment; and marked (grade 4), >51% of hepatocytes contained pigment and the pigment was very dense. Hepatocellular necrosis was characterized by multiple adjacent hepatocytes that were swollen with increased eosinophilia, karyorrhectic nuclear debris, with or without accompanying inflammatory cells. A four-grade severity scale was used: minimal (grade 1), up to three focal areas of necrosis present; mild (grade 2), necrosis in more than three involved regions or up to 25% of the liver; moderate (grade 3), necrosis in 26% to 60% of the liver; and severe (grade 4), necrosis in >61% of the liver.
Eosinophilic and basophilic hepatocellular foci were diagnosed when there was an enlargement of the hepatocytes with increased acidophilic or basophilic staining, respectively, compared with the surrounding normal liver cells. Foci typically had a discrete lesion margin, where attenuated hepatocytes at the lesion margin (compression) involved <70% of the lesion circumference. There was preservation of lobular architecture and absence of cellular atypia. The distinction between large foci (usually eosinophilic or mixed) and hepatocellular adenomas was made on the basis of retention of normal lobular architecture in the foci, greater size of hepatocellular adenomas (usually measuring at least 3 mm), and presence of compression or bulging from the liver surface along >70% of the lesion circumference.
Bile duct hyperplasia was diagnosed when increased numbers of small bile ducts arose in portal regions. The biliary epithelium lining the ducts was well differentiated, forming normal ducts.
Pancreas: In male rats, there were significant increases in the incidences of pancreatic acinar adenoma and pancreatic acinar adenoma or carcinoma (combined) in the 3,000 and 10,000 ppm groups compared to the control group (Table 14). In males, acinar carcinomas were present in the 3,000 and 10,000 ppm groups, but the incidences were not significant (Table 14). In females, acinar adenomas were observed in the 3,000 and 10,000 ppm groups, but the incidences were not significant (Table 14). Pancreatic acinar adenomas were characterized as distinct nodular masses that were not contiguous with the adjacent parenchyma, which were >3 mm in diameter, and that compressed the adjacent tissue; pleomorphism or atypia was rarely present. Pancreatic acinar carcinomas were typically larger than adenomas and frequently exhibited cellular pleomorphism and atypia; invasion or metastasis was pathognomonic. Scirrhous reactions were occasionally present, characterized by dense fibrous or connective tissue. Pancreatic acinar hyperplasias were higher in the 1,000, 3000, and 10,000 ppm male groups and in the 3,000 and 10,000 ppm female groups compared to the control group, but the differences were not significant.
There was a clear morphological continuum from focal acinar hyperplasia to adenoma and to carcinoma. Pancreatic acinar hyperplasia was characterized by circumscribed areas of enlarged acini that were <3 mm in diameter and that were contiguous with the adjacent parenchyma. Severity grades were assigned using a four-grade scale: minimal (grade 1), no more than one lobule was affected and the lesion was smaller than 1 mm; mild (grade 2), lesion was 1 to 2 mm; moderate (grade 3), lesion was 2 to 3 mm; and marked (grade 4), lesion was 3 mm but lacked features of an adenoma, such as compression. Severity grades were increased if multiple lesions were present.
Gross lesions of the reproductive tract: Significantly increased incidences of several genitourinary abnormalities consistent with perturbation of developmental testosterone signaling and Wolffian duct differentiation were evident among 10,000 ppm DEHP-exposed males relative to control animals (Table 15). Small reproductive tissues were noted in 10,000 ppm males, including, but not limited to, the phallus, testes, epididymides, seminal vesicles, levator ani/bulbocavernosus (LABC) muscle, Cowper’s glands, and prostate (Table 15; Appendix H). Findings in the testes (small) were particularly prevalent, occurring in all 10,000 ppm males that survived past the initial 2 weeks of the 2-year study period relative to only two incidences in examined control rats (Table 15). Undescended testes (unilateral or bilateral) were noted in the abdomen in 19 (approximately 39%) rats and the inguinal region in 4 (approximately 8%) rats in the 10,000 ppm group relative to a single incidence of undescended testes in the abdomen (bilateral) in control males. Findings commonly associated with testicular retention in the abdomen, such as the presence of a cranial suspensory ligament (approximately 10%) and extended (>20 mm; approximately 16%) or absent gubernaculum (unilateral or bilateral; approximately 35%), were also significantly increased in the 10,000 ppm males relative to control animals (Table 15). Exposure-related increases in the left and right mean gubernaculum lengths were evident in 10,000 ppm males. Using a nonparametric (rank-based) analysis, a significant increase in the length of the right gubernaculum was observed in 10,000 ppm males. This corresponded to data in which the effect was largely associated with increased right gubernaculum lengths (≥35 mm) in seven males in the 10,000 ppm group compared to five animals with these lengths in the left gubernaculum (Table 15; Appendix H). In addition, there were 18 animals with gubernaculum not present, 12 of which had a bilateral finding. Epididymal anomalies, such as agenesis, are typically seen concomitant with other abnormalities in organs that are developed from Wolffian ducts. Agenesis of all or part of the epididymis (unilateral or bilateral) was observed only in rats in the 10,000 ppm group. Incomplete separation of the prepuce (approximately 14%), cleft phallus (approximately 6%), and/or cleft prepuce (2%) was observed in 10,000 ppm males relative to no incidences in control rats (Table 15).
Limited genitourinary abnormalities were associated with DEHP exposure in female rats. A greater incidence of vaginal nonpatency (approximately 10%) occurred in 10,000 ppm females; in contrast a similar finding was not found in any other exposure group. Additionally, observations of cleft phallus were noted in both the 3,000 ppm (approximately 4%) and 10,000 ppm (approximately 2%) females.
Testis: There were significant increases in germinal epithelium degeneration (includes bilateral), interstitial cell hyperplasia (includes bilateral), and seminiferous tubule dysgenesis (includes bilateral) in the 10,000 ppm group compared to the control group (Table 16). Two of the occurrences of seminiferous tubule dysgenesis were bilateral (Table 16; Appendix H). Germinal epithelium degeneration was recorded when one or more of the following features was present in tubules not adjacent to the rete testis: tubular vacuolation, partial depletion of germ cells, degenerating (multinucleated or apoptotic) germ cells, disordered arrangement of the germ cell layers, or seminiferous tubules completely devoid of germ cells (atrophy) and lined only by Sertoli cells. Germinal epithelium degeneration was scored for severity on a four-grade scale: minimal (grade 1), up to 25% of at least one testis involved; mild (grade 2), 26% to 50% of at least one testis involved; moderate (grade 3), 51% to 75% of at least one testis involved; and marked (grade 4), rare to no normal seminiferous tubules in either testis were present (i.e., remaining seminiferous tubules solely lined by Sertoli cells).
Interstitial cell hyperplasia, defined as focal aggregates of Leydig cells, was scored using a four-grade scale: minimal (grade 1), when only a thin rim of interstitial cells or a cluster of cells one-fourth the size of a normal seminiferous tubule was present; mild (grade 2), when several such areas were present or one cluster was present that was one-half the size of a normal seminiferous tubule; moderate (grade 3), when a cluster three-fourths the size of a normal seminiferous tubule was present; and marked (grade 4), when a cluster of interstitial cells approached the diameter of a normal seminiferous tubule. The interstitial cells involved were frequently elongated and flattened in profile. Interstitial cell adenomas were characterized by regions of increased interstitial cells, described as mostly uniform polyhedral cells with abundant eosinophilic, finely granular, or vacuolated cytoplasm, which exceeded the diameter of three seminiferous tubules. Circumferential compression of adjacent seminiferous tubules was observed occasionally.
Seminiferous tubule dysgenesis only occurred in the highest exposure concentration group (10,000 ppm) and was characterized by seminiferous tubules that were misshapen and contorted, lined by only Sertoli cells, surrounded by a thickened basement membrane, and often accompanied by Leydig cell aggregates (Figure 6). The dysgenesis lesions were commonly associated with an undescended testis but were also identified in scrotal testes. Three of the 10 animals with seminiferous tubule dysgenesis died early, each on day 3 of the study (two were 24 days old and one was 23 days old), and all seminiferous tubule dysgenesis lesions were focal. Focal lesion locations were variable, sometimes being present in sections that did not include the rete testis.
Epididymis: There were significant increases in the incidences of epididymis hypospermia (includes bilateral) in the 10,000 ppm group compared to the control group (Table 16). Epididymis hypospermia was characterized by a reduced density of sperm in the lumen of the epididymal duct, often accompanied by luminal cell debris. It was scored using a four-grade scale: minimal (grade 1), 25% to 50% reduction of spermatozoa; mild (grade 2), 51% to 66% reduction; moderate (grade 3), 67% to 80% reduction; and marked (grade 4), 81% to 100% reduction.
Uterus: A positive trend with increasing exposure concentration in uterus endometrium adenocarcinoma and uterus adenoma, adenocarcinoma, squamous cell carcinoma, or squamous cell papilloma (combined) (Table 17). Uterus endometrium adenocarcinoma typically is poorly circumscribed and invades the myometrium. The neoplastic epithelial cells form solid nests, cords, papillary, or acinar structures that are within, or supported by, stroma.
Kidney: There were significant increases in the incidences of nonneoplastic kidney lesions in male and female DEHP-exposed groups relative to the control groups (Table 18). The incidences of papilla edema and papilla epithelium hyperplasia were significantly increased in the 10,000 ppm males and females relative to control animals; there was a significant increase in the incidence of papilla hemorrhage in the 10,000 ppm males. There were significant increases in the incidence of kidney infarct in the 300 and 10,000 ppm males and the 1,000 and 10,000 ppm females. A significant increase in the incidence of renal tubule cyst was observed in the 10,000 ppm female group compared to that of the control group. A positive trend in the incidence of renal tubule dilation occurred with increasing exposure concentration in the females.
Papilla edema (Figure 7) occurred only in exposed animals, was present in most males and females exposed to 10,000 ppm, and was observed in two females exposed to 1,000 ppm DEHP. Papillary edema affected the kidneys bilaterally and was characterized by expansion of the papillary interstitium by fibrillary amphophilic to pale eosinophilic material. Collecting ducts were often dilated and distorted and lined by a continuous layer of thin attenuated epithelium. The dilatation of collecting ducts sometimes extended to include renal tubules within the medulla and/or cortex. Representative sections were stained with Alcian blue, which identifies glycosaminoglycans (GAGs), and with periodic acid-Schiff (PAS) to evaluate basement membrane integrity. The interstitial material stained positively for Alcian blue, confirming that the material contained GAGs. The basement membranes of vascular structures stained intensely positive for PAS throughout the kidney sections and were not compromised in any areas, indicating they were intact. The renal tubules in the cortex and outer medulla stained uniformly and intensely positive for PAS, but there was an abrupt loss of staining at the junction of the outer and inner medulla. Tubules and collecting ducts within the inner medulla and papilla lacked staining for PAS, indicating disruption of the basement membrane of tubules/ducts within this region.
Papillary hemorrhage occurred in regions of papillary edema (Figure 8). Papilla epithelium hyperplasia was diagnosed when there was thickening and/or variably sized outgrowths (with clear spaces) of the epithelium overlying the renal papilla (Figure 9). Occasionally, these spaces contained eosinophilic material or cells. Although papillary epithelial hyperplasia is commonly associated with advanced chronic progressive nephropathy (CPN), there was no direct correlation with CPN severity in this study.
Kidney infarct consisted of well-demarcated, wedge-shaped regions characterized by renal interstitial fibrosis and depression of the overlying capsule; lesions extended from the capsular surface into the medulla. Infarcts were scored using a four-grade severity scale: minimal (grade 1), <25% of renal involvement; mild (grade 2), 25% to 50% of renal involvement; moderate (grade 3), 51% to 75% of renal involvement; and marked (grade 4), >75% renal involvement. Renal tubule cysts were characterized by dilated renal tubules lined by cuboidal to thin attenuated epithelial cells. There was a positive trend in renal tubule dilation in females.
Heart: A significant increase in the incidences of heart valve fibrosis and heart valve thrombus occurred in the 10,000 ppm males, relative to the control animals (Table 19). Heart valve fibrosis was diagnosed when valves were expanded by fibrous connective tissue that was more densely eosinophilic than the loose lightly basophilic to amphophilic tissue of a normal heart valve (Figure 10). Severity was scored using a four-grade scale: minimal (grade 1), change was barely detectable to thickening of the valve up to double the normal thickness; mild (grade 2), thickening up to three times the normal thickness, involvement of several portions of the valve, or more than one valve up to double the normal thickness; moderate (grade 3), thickening of several regions of the valve or valves, at least one of which was greater than double the normal thickness of a valve at that location; marked (grade 4) valve fibrosis was not diagnosed. Heart valve thrombus was characterized by fibrin, admixed with variable numbers of blood cells, that covered the cardiac valves. Severity grades were assigned according to the following grading scheme: minimal (grade 1), amount of fibrin and cells was less than the thickness of the widest part of the valve; mild (grade 2), layering upon the valves that could occlude approximately 50% of the valve lumen; moderate (grade 3), lesions that occluded 51% to 80% of the valve lumen; marked (grade 4), lesions that occluded ≥81% of the valve lumen.
Bone Marrow: There was a significant increase in the incidence of bone marrow hypercellularity in the 3,000 and 10,000 ppm male groups, relative to the control group (Table 19). Bone marrow hypercellularity was characterized by an increase in one or more hematopoietic cell lines, generally with a decrease in adipocytes.
Pituitary Gland: There was a significant increase in the incidence of pars distalis hypertrophy in the 3,000 and 10,000 ppm males compared to control animals (Table 19). Pars distalis hypertrophy was characterized by clusters of cells that were round, with abundant vacuolated amorphous amphophilic or pale eosinophilic cytoplasm and peripherally compressed nuclei, scattered throughout the pars distalis (Figure 11). A severity grade was assigned based on the numbers of affected cells.
The toxicological significance of other lesion findings is unknown as the response was considered either marginal, not dose-responsive, and/or potentially decreased (potentially due to lower body weights) (Appendix H). In males, these lesions included: adrenal cortex focal hyperplasia, adrenal medulla focal hyperplasia, testis polyarteritis nodosa, bilateral testis polyarteritis nodosa, thyroid gland C-cell adenoma, and thyroid gland C-cell adenoma or carcinoma (combined). In females, these lesions included: nose respiratory epithelium hyaline droplet accumulation, ovary atrophy, uterus endometrium squamous metaplasia, mammary gland fibroadenoma, pituitary gland pars distalis, or unspecified site adenoma in females.
Postweaning-only Study in Rats (Study 2)
Overall, survival at study termination of male and female rats exposed to DEHP was commensurate with or greater than that of control animals (Table 20; Figure 12). Survival to study termination was significantly increased in 10,000 ppm males (approximately 84%) relative to control males (approximately 64%).
At study termination, group mean body weights for the 300, 1,000, and 3,000 ppm DEHP groups were within 6% of control animals in both male and female rats (Table 21, Table 22; Figure 13). Lower body weights were noted in males (approximately 16%) and females (approximately 22%) in the 10,000 ppm groups at the end of study relative to control animals. These effects were attributed to reduced body weight gain relative to control animals, which occurred throughout the study.
Feed consumption by male and female rats in the 300, 1,000, 3,000, and 10,000 ppm DEHP groups was commensurate with that of the control group throughout the study with the exception of study week 1, when feed consumption was approximately 21% lower in the 10,000 ppm male and female groups (Table 23, Table 24). This finding might reflect an initial adjustment related to the palatability of feed containing high concentrations (1%) of DEHP. Dietary concentrations of 300, 1,000, 3,000, and 10,000 ppm resulted in average daily doses of approximately 17, 54, 170, and 602 mg/kg/day for males and 17, 60, 177, and 646 mg/kg/day for females (Appendix H).
No exposure-related clinical findings were observed in any of the exposed groups (Appendix H).
Pathology
This section describes the statistically significant or biologically noteworthy changes in the incidences of neoplasms and/or nonneoplastic lesions of the liver, pancreas, male and female reproductive organs, heart, bone marrow, and pituitary gland. Summaries of the incidences of neoplasms and nonneoplastic lesions, individual animal neoplasm diagnoses, and statistical analyses of primary neoplasms that occurred with an incidence of at least 5% in at least one animal group are presented as supplemental data in Appendix H.
Liver: There were significant increases in the incidences of hepatocellular adenomas (10,000 ppm males and females) and carcinomas (10,000 ppm males) relative to that of control animals and a positive trend in females for hepatocellular carcinomas with increasing exposure concentration (Table 25). The incidence of adenoma or carcinoma (combined) was significantly increased in the 10,000 ppm male and female groups relative to the respective control groups. Hepatocellular adenomas were characterized by regions that were sharply demarcated from surrounding liver parenchyma, nodular, and compressing adjacent normal hepatocytes, with loss of normal lobular architecture and an irregular growth pattern. The liver plates typically impinged obliquely on the surrounding liver parenchyma. The hepatocytes within an adenoma generally varied in size. Hepatocellular carcinomas were characterized by one or more of the following features: local infiltrating growth and/or distinct lack of demarcation with the adjacent tissue, the presence of trabeculae composed of multiple layers of hepatocytes, cellular pleomorphism, loss of normal lobular architecture, regions of hemorrhage and/or necrosis, and increased mitotic figures.
There were significant increases in the incidences of many nonneoplastic liver lesions in DEHP-exposed groups relative to the control groups (Table 25). The incidence of hepatocellular cytoplasmic alteration was significantly increased in 3,000 and 10,000 ppm males and in 1,000, 3,000, and 10,000 ppm females relative to that of the respective control animals. There were significant increases in the incidence of hepatocellular hypertrophy in the 10,000 ppm males and in the 1,000, 3,000, and 10,000 ppm females. Significant increases in the incidence of liver pigment were observed in the 1,000, 3,000, and 10,000 ppm males and females. There were significant increases in the incidence of liver necrosis in the 3,000 and 10,000 ppm males. There was a significant increase in the incidence of hepatocellular eosinophilic foci in the 10,000 ppm males, and a positive trend in the incidence of hepatocellular clear cell foci in exposed males with increasing exposure concentration.
Hepatocellular cytoplasmic alteration was characterized by hepatocytes that were expanded with eosinophilic granular cytoplasm (see Figure 5 as an example). A four-grade severity scale was used based on degree of tissue affected in the section of liver that was evaluated histologically: minimal (grade 1), up to 25% of hepatocyte involvement; mild (grade 2), 26% to 50% of hepatocyte involvement; moderate (grade 3), 51% to 75% of hepatocyte involvement; and marked (grade 4) at least 76% of hepatocyte involvement. Hepatocellular hypertrophy often occurred in conjunction with cytoplasmic alteration and/or pigment. Hypertrophy was characterized by enlargement of the hepatocytes. In lesser affected animals, hypertrophy was confined to centrilobular regions, but in more severely affected animals, hypertrophy extended into the midzonal and periportal areas. A four-grade severity scale was used: minimal (grade 1), up to 10% of hepatocyte involvement; mild (grade 2), 11% to 25% of hepatocyte involvement; moderate (grade 3), 26% to 50% of hepatocyte involvement; and severe (grade 4), ≥51% of hepatic involvement. Hypertrophy was generally centrilobular and often involved only a few cells per lobule. Although hypertrophy was only occasionally observed in males (at the 3,000 and 10,000 ppm concentrations), in females, its incidence (but not severity) increased significantly with exposure concentrations starting at 1,000 ppm.
Pigment was characterized by a pale gold-colored pigment within the hepatocellular cytoplasm (see Figure 5 as an example). A four-grade severity scale was used: minimal (grade 1), up to 30% of hepatocytes contained pigment; mild (grade 2), 31% to 50% of hepatocytes contained pigment; moderate (grade 3), ≥51% of hepatocytes contained pigment; and marked (grade 4), >51% of hepatocytes contained pigment, and the pigment was very dense. Hepatocellular necrosis was characterized by multiple adjacent hepatocytes that were swollen with increased eosinophilia, karyorrhectic nuclear debris, with or without accompanying inflammatory cells. Necrosis was scored using a four-grade severity scale: minimal (grade 1), up to three focal areas of necrosis present; mild (grade 2), necrosis in up to 25% of the liver; moderate (grade 3), necrosis in 26% to 60% of the liver; and severe (grade 4), necrosis in ≥61% of the liver.
Hepatocellular foci were diagnosed when there was an alteration in the arrangement of hepatocytes involving at least six cells, with a discrete lesion margin, where attenuated hepatocytes at the lesion margin (compression) involved <70% of the lesion circumference. Lobular architecture was preserved in the absence of cellular atypia, and the hepatocytes within the eosinophilic foci were more eosinophilic than those within the surrounding parenchyma. Clear cell foci were characterized by circular or ovoid regions composed of normal-sized or enlarged cells with distinct cytoplasmic clear spaces compared with the surrounding parenchyma. The nuclei were often small and dense, prominent, and centrally located, occasionally exhibiting increased volume. The distinction between large foci (usually eosinophilic) and hepatocellular adenomas was based on retention of normal lobular architecture in the foci, greater size of hepatocellular adenomas (usually measuring at least 3 mm), and presence of compression or bulging of the adenoma from the liver surface along >70% of the lesion circumference.
Pancreas: In male rats, there were significant increases in the incidences of acinar adenoma and acinar adenoma or carcinoma (combined) in the 3,000 and 10,000 ppm groups relative to the control group. There was a significant increase in the incidence of acinar carcinoma in the 10,000 ppm male group (Table 26). In females, there was a positive trend for pancreas acinar adenoma or carcinoma (combined). Pancreatic acinar adenomas were distinct nodular masses that were not contiguous with the adjacent parenchyma, which were >3 mm in diameter, and that compressed the adjacent tissue; pleomorphism or atypia was rarely present. Pancreatic acinar carcinomas were typically larger than adenomas and frequently exhibited cellular pleomorphism and atypia; invasion or metastasis was pathognomonic. Scirrhous reactions were occasionally present, characterized by dense fibrous or connective tissue.
There were significant increases in the incidences of pancreatic acinus hyperplasia in male rats in the 3,000 and 10,000 ppm groups relative to the control group. In females, there was a significant increase in the incidence of pancreatic acinus hyperplasia in the 10,000 ppm group. Pancreatic acinus hyperplasia was characterized by circumscribed areas of enlarged acini that were <3 mm in diameter and that were contiguous with the adjacent parenchyma. A four-grade severity scale was used: minimal (grade 1), no more than one lobule was affected, and the lesion was smaller than 1 mm; mild (grade 2), lesion was 1 to 2 mm; moderate (grade 3), lesion was 2 to 3 mm; and marked (grade 4), lesion was 3 mm but lacked features of an adenoma, such as compression. Severity grades were increased if multiple hyperplastic lesions were present within the pancreas.
Testis: There was a positive trend in the incidence of testicular interstitial cell adenoma in male rats (Table 27). Interstitial cell adenomas were characterized by regions of increased Leydig cells, described as mostly uniform polyhedral cells with abundant eosinophilic, finely granular, or vacuolated cytoplasm, which exceeded the diameter of three seminiferous tubules. Circumferential compression of adjacent seminiferous tubules was observed occasionally.
There were significant increases in the incidences of germinal epithelium degeneration, bilateral germinal epithelium degeneration, testis edema, and bilateral testis edema in the 10,000 ppm group compared to the control males; there was a positive trend in the incidence of focal interstitial cell hyperplasia (Table 27). Germinal epithelium degeneration was recorded when one or more of the following features was present in tubules not adjacent to the rete testis: tubular vacuolation, partial depletion of germ cells, degenerating (multinucleated or apoptotic) germ cells, and disordered arrangement of the germ cell layers and/or seminiferous tubules completely devoid of germ cells and lined only by Sertoli cells. Germinal epithelium degeneration was scored using a four-grade severity scale: minimal (grade 1), up to 25% of at least one testis involved; mild (grade 2), 26% to 50% of at least one testis involved; moderate (grade 3), 51% to 75% of at least one testis involved; and marked (grade 4), rare to no normal seminiferous tubules in either testis were present (i.e., remaining seminiferous tubules solely lined by Sertoli cells).
Testis edema was characterized by the presence of acellular, finely granular or fibrillar pale eosinophilic material in the interstitium. In most animals, this finding was bilateral. A severity grade was determined by the amount of interstitial fluid. Generally, the severity of edema was higher in testes with reduced numbers of seminiferous tubules, because the interstitial fluid filled the intervening space. In some animals with reduced numbers of seminiferous tubules, however, the testis was collapsed and shrunken, leaving no interstitial space.
Interstitial cell hyperplasia was scored using a four-grade severity scale: minimal (grade 1), when only a thin rim of interstitial cells or a cluster of cells one-fourth the size of a normal seminiferous tubule was present; mild (grade 2), when several such areas were present or one cluster was present that was one-half the size of a normal seminiferous tubule; moderate (grade 3), when a cluster three-fourths the size of a normal seminiferous tubule was present; and marked (grade 4), when a cluster of interstitial cells approached the diameter of a normal seminiferous tubule. The interstitial cells involved were frequently very elongated and flattened in profile.
Epididymis: There were significant increases in the incidences of bilateral hypospermia and bilateral epididymis duct exfoliated germ cell in the 10,000 ppm group relative to the control males (Table 27). Epididymis hypospermia was characterized by a reduced density of sperm in the lumen of the epididymal duct, often accompanied by luminal cell debris. Its severity was scored using a four-grade scale: minimal (grade 1), 25%–50% reduction of spermatozoa; mild (grade 2), 51%–66% reduction; moderate (grade 3), 67%–80% reduction; and marked (grade 4), 81%–100% reduction. The lesion of epididymis duct exfoliated germ cell was characterized by the presence of nondegenerate germ cells and debris in the epididymal lumen. This was often accompanied by depletion of germ cells from the seminiferous epithelium in testes diagnosed with germinal epithelium degeneration.
Uterus: There was a significant increase in the incidence of endometrium adenocarcinoma in the 10,000 ppm group and a positive trend in the incidence of uterine squamous cell papilloma with increasing exposure concentration (Table 28). Uterus adenocarcinomas were typically poorly circumscribed and invaded the myometrium. The neoplastic epithelial cells formed solid nests, cords, papillary, or acinar structures. Uterine squamous cell papillomas were characterized by a neoplasm that arose from the surface epithelium with either a broad base or a delicate stalk. The epithelium was well differentiated and arranged in papillary, glandular, or tubular structures that were lined by cuboidal to columnar cells, one to two cell layers thick. The combined incidence of these was significantly increased in the 10,000 ppm group (Table 28).
There were significant increases in the incidences of uterine inflammation in the 300, 1,000, and 10,000 ppm groups, compared to the control group (Table 28). Uterine inflammation was characterized by a spectrum of changes from mostly mononuclear cells (recorded as chronic inflammation) to a mixture of mononuclear cells and neutrophils (recorded as chronic active inflammation); both diagnoses were considered a part of the same process.
Heart: There were significant increases in the incidences of heart valve fibrosis and heart valve thrombus in the 10,000 ppm male group relative to the control group (Table 29). Valve fibrosis was diagnosed when valves were expanded by fibrous connective tissue that was more densely eosinophilic than the loose lightly basophilic to amphophilic tissue of a normal heart valve. Heart valve thrombus was characterized by fibrin, admixed with variable numbers of blood cells, which covered the cardiac valves.
Bone Marrow: There was a significant increase in the incidence of bone marrow hypercellularity in the 1,000 and 10,000 ppm male groups relative to the control group (Table 29). Bone marrow hypercellularity was characterized by an increase in one or more hematopoietic cell lines, generally with a decrease in adipocytes.
Pituitary Gland: There was a significant increase in the incidence of pars distalis hypertrophy in the 10,000 ppm males compared to the control group (Table 29). Pars distalis hypertrophy was characterized by clusters of cells that were enlarged, with abundant amorphous amphophilic or pale eosinophilic cytoplasm and peripherally compressed nuclei (“signet ring” cells). A severity grade was assigned based on the numbers of affected cells.
The toxicological significance of other lesion findings is unknown as the response was considered either marginal, not dose-responsive, and/or decreased (potentially due to lower body weights). (Appendix H). In males, these lesions included heart cardiomyopathy and parathyroid gland diffuse hyperplasia. In females, these lesions included: heart cardiomyopathy; uterus endometrium metaplasia; mammary gland fibroadenoma; mammary gland fibroma, fibroadenoma, or adenoma; mammary gland fibroma, fibroadenoma, carcinoma, or adenoma; nose, olfactory epithelium, hyaline droplet accumulation; pituitary gland pars distalis adenoma; thyroid gland C-cell hyperplasia; and thyroid gland C-cell adenoma or carcinoma.
Comparative Carcinogenic Benchmark Dose Analysis
Exposure-related neoplastic lesions were further assessed via benchmark dose (BMD) analyses. Daily doses for each exposure group were calculated using time-weighted averages of postweaning feed consumption and corresponding chemical intake during the 2-year exposure period for each study. All available dichotomous models in U.S. EPA’s BMD Software (BMDS version 3.1.2)169 were fit to the adjusted incidence data for assessed neoplastic lesions. Model fit was assessed by a chi-square goodness-of-fit test, visual inspection of the respective plots of observed versus predicted values from the various models, and Akaike information criterion (AIC) values (Appendix F). A benchmark response (BMR) of 0.1, corresponding to a 10% extra risk of a DEHP carcinogenic response, was used to determine benchmark doses. Benchmark doses (i.e., BMD10 [BMD corresponding to a 10% extra risk] and BMDL10 [95% lower bound on the BMD corresponding to a 10% extra risk]) were determined for incidences of hepatocellular adenoma or carcinoma (combined), pancreatic acinar adenoma or carcinoma (combined), testicular interstitial cell adenoma, and uterine (including cervix) adenoma, adenocarcinoma, squamous cell carcinoma, or squamous cell papilloma (combined). The BMD10 and BMDL10 were calculated separately for the perinatal and postweaning study (Study 1) and for the postweaning-only study (Study 2).
Higher adjusted incidences of hepatocellular adenoma or carcinoma (combined) occurred in the 3,000 and 10,000 ppm male rats exposed during the perinatal and postweaning periods (6.7% and 30.6%), relative to postweaning-only exposure (2.2% and 25.6%) (Table 30). A probit model provided the best relative model fit for the adjusted rates of hepatocellular adenoma or carcinoma (combined) in the perinatal and postweaning study (Figure 14A). Using this model, a BMD10 of 382.90 mg/kg/day was estimated for hepatocellular adenoma or carcinoma (combined) in male rats. A multistage degree 4 model provided the best relative model fit for the adjusted rates of hepatocellular adenoma or carcinoma (combined) in the postweaning-only study (Figure 14B). Using this model, a BMD10 of 434.41 mg/kg/day was estimated for hepatocellular adenoma or carcinoma (combined) observed in male rats.
A higher adjusted incidence of hepatocellular adenoma or carcinoma (combined) occurred in 3,000 ppm female rats exposed during the perinatal and postweaning periods (20.9%) relative to postweaning-only exposure (2.3%) (Table 31). A log-logistic model provided the best relative model fit for the adjusted rates of hepatocellular adenoma or carcinoma (combined) in the perinatal and postweaning study (Figure 15A). Using this model, a BMD10 of 122.95 mg/kg/day was estimated for hepatocellular adenoma or carcinoma (combined) in female rats. A multistage degree 4 model provided the best relative model fit for the adjusted rates of hepatocellular adenoma or carcinoma (combined) in the postweaning-only study (Figure 15B). Using this model, a BMD10 of 383.63 mg/kg/day was estimated for hepatocellular adenoma or carcinoma (combined) in female rats.
A higher adjusted incidence of pancreatic acinar adenoma or carcinoma (combined) occurred in 3,000 ppm male rats exposed during the perinatal and postweaning periods (81.2%), relative to postweaning-only exposure (49.9%); however, incidences in the 10,000 ppm group were similar between the two studies (62.5% versus 69.8%, respectively) (Table 30). A dichotomous Hill model provided the best relative model fit for the adjusted rates of pancreatic acinar adenoma or carcinoma (combined) in the perinatal and postweaning study (Figure 16A). Using this model, a BMD10 of 85.92 mg/kg/day was estimated for pancreatic acinar adenoma or carcinoma (combined) in male rats (Table 30). A log-logistic model provided the best relative model fit for the adjusted rates of pancreatic acinar adenoma or carcinoma (combined) in the postweaning-only study (Figure 16B). Using this model, a BMD10 of 30.99 mg/kg/day was estimated for pancreatic acinar adenoma or carcinoma (combined) in male rats (Table 30).
The incidences of pancreatic acinar adenoma or carcinoma (combined) in female rats were not amenable to BMD modeling. These lesions are considered rare in female rats and were only observed at adjusted rates up to 5% in any single exposed group across both studies. Therefore, an estimated BMR corresponding to 10% extra risk would be greater than the maximum exposure concentration used in the study.
A higher adjusted incidence of testicular interstitial cell adenoma occurred only in 10,000 ppm male rats exposed during the postweaning period (32.3%), relative to perinatal and postweaning exposure (14.1%) (Table 16, Table 27, Table 32). Although there was no exposure-related response in the testis from the perinatal and postweaning study (Study 1), there was a marginal response in the testis with postweaning-only exposure (Study 2). A multistage degree 4 model provided the best relative model fit for the adjusted rates of testicular interstitial cell adenoma in the postweaning-only study (Table 32; Figure 16; Appendix H). Using this model, a BMD10 of 366.69 mg/kg/day was estimated for testicular interstitial cell adenoma in male rats.
Higher adjusted incidences of uterine (including cervix) adenocarcinoma, adenoma, squamous cell carcinoma, or squamous cell papilloma (combined) occurred in 3,000 and 10,000 ppm female rats exposed postweaning-only (13.4% and 30.7%, respectively), relative to exposure during the perinatal and postweaning periods (7% and 19%, respectively) (Table 31). A logistic model provided the best relative model fit for the adjusted rates of uterine neoplasms in the perinatal and postweaning study (Figure 17A). Using this model, a BMD10 of 594.19 mg/kg/day was estimated for uterine (including cervix) adenocarcinoma, adenoma, squamous cell carcinoma, or squamous cell papilloma (combined) in female rats (Table 31). A probit model provided the best relative model fit for the adjusted rates of uterine (including cervix) adenocarcinoma, adenoma, squamous cell carcinoma, or squamous cell papilloma (combined) in the postweaning-only study (Figure 17B). Using this model, a BMD10 of 324.15 mg/kg/day was estimated for uterine (including cervix) adenocarcinoma, adenoma, squamous cell carcinoma, or squamous cell papilloma (combined) in female rats (Table 31).
Genetic Toxicology
DEHP was tested in a variety of genotoxicity assays in vitro and in vivo; most results were negative. DEHP (100–10,000 µg/plate) was tested in six independent bacterial mutation assays using a variety of strains of Salmonella typhimurium (TA100, TA1535, TA1537, TA97, and TA98) and exogenous metabolic activation systems (induced hamster, rat, or mouse liver S9 plus cofactors); results from all bacterial assays were negative (Appendix H).149 A single mouse lymphoma gene mutation assay was conducted with DEHP (0.125–3.0 µL/mL) and was negative overall, with and without induced rat liver S9 mix (Appendix H). In three independent studies, no increases in chromosomal aberrations were observed in Chinese hamster ovary (CHO) cells exposed to DEHP (concentrations up to 5,000 µg/mL) with or without induced rat liver S9 (Appendix H).153 In a series of nine in vitro sister chromatid exchange (SCE) tests conducted in CHO cells with and without S9, DEHP produced positive responses in four tests, equivocal results in three, and negative results in two (Appendix H).153 All of the increases in SCEs judged to be positive or equivocal were observed only in the absence of S9 and at concentrations of DEHP that induced severe cell cycle delay, necessitating longer incubation prior to cell harvesting. The level of cytotoxicity and the extended incubation times might have contributed to the increased SCE levels observed in these seven studies, rather than the SCE reflecting a direct interaction of DEHP with chromosomal DNA. DEHP was tested for induction of sex-linked recessive lethal mutations in male Drosophila melanogaster in two independent studies, one using adult injection and one using larval feeding as the route of exposure; both studies yielded negative results (Appendix H).158,159
In vivo, no significant increases in chromosomal aberrations were observed in bone marrow cells of female B6C3F1 mice following exposure to DEHP (3,000–12,000 ppm) in dosed feed for 14 days (Table D-1). DEHP was tested in three independent erythrocyte micronucleus tests and produced varying results. In one test, B6C3F1 female mice were exposed to DEHP (3,000–12,000 ppm) in dosed feed for 14 days; results were judged to be equivocal overall—the response was negative in the immature erythrocyte population (polychromatic erythrocytes) and positive in the mature erythrocyte population (normochromatic erythrocytes) (Table D-2). In a second test, DEHP (1,500–6,000 ppm) induced an equivocal response in male TgAC (FVB/N) mice and a positive response in female TgAC (FVB/N) mice following exposure via dosed feed for 26 weeks (Table D-3). Another 26-week exposure test in TgAC (FVB/N) mice used dermal application of DEHP (100–400 mg/kg/day) and produced negative results in both male and female mice (Table D-4).
Discussion
Di(2-ethylhexyl) phthalate (DEHP) is a plasticizer to which humans are exposed, as evidenced by detection of DEHP metabolites in serum and urine samples. The presence of DEHP metabolites in human amniotic fluid samples shows exposure occurs in utero. Rodent studies report that DEHP produces adverse effects on the developing male rat reproductive tract and induces hepatic, pancreatic, and testicular neoplasms. Prior to the current studies, data were insufficient to assess whether developmental exposure would alter lifetime DEHP carcinogenic risk. To address this knowledge gap, the National Toxicology Program (NTP) conducted two 2-year bioassays with DEHP administered in feed to Sprague Dawley (Hsd:Sprague Dawley SD) rats to evaluate whether neoplasm incidence during lifetime exposure that included the perinatal period (gestation and lactation) would increase the incidence of neoplasms or lead to the appearance of different neoplasm types relative to chronic exposure initiated in early adulthood.
In the perinatal and postweaning study (Study 1), exposure was associated with significantly decreased maternal mean body weights during gestation and lactation in the 10,000 ppm group compared to the control group, with the magnitude of the effect increasing throughout the perinatal period. This effect was attributed to significantly decreased body weight gain during gestation, likely in part due to cumulative effects of reduced maternal feed consumption (g/animal/day) throughout the gestation and lactation period in the 10,000 ppm group.
Estimated DEHP intake increased in proportion to exposure concentration, with the exception of 10,000 ppm dams during lactation (lactation days [LDs] 1–14), for which significantly decreased feed consumption resulted in a less-than-proportional higher intake. In gestation day (GD) 18 dams, the mean concentration of the DEHP metabolite, mono(2-ethylhexyl) phthalate (MEHP), increased with exposure concentration although the increase was more than proportional (63-fold increase in plasma concentration versus 30-fold increase in DEHP intake [mg DEHP/kg body weight/day or mg/kg/day] during gestation from lowest [300 ppm] to highest [10,000 ppm] exposure groups). Amniotic fluid and fetus concentrations of MEHP increased 23- and 46-fold, respectively, from the lowest to highest exposure groups (300 to 10,000 ppm). Gestational transfer of MEHP from the dam to the fetus was moderate.
MEHP concentrations in dam plasma on GD 18 at the lowest exposure concentration were 630 ng/mL, approximately 60-fold higher than the median blood MEHP concentration of 10.4 ng/mL observed in pregnant women in the Hokkaido Study Sapporo Cohort.203 Moreover, the GD 18 MEHP concentration measured in amniotic fluid at the lowest exposure concentration was 73.4 ng/mL, which is 28-fold higher than the upper 95th percentile of MEHP levels measured in human amniotic fluid samples.26 Additional studies have detected MEHP in human amniotic fluid and cord blood plasma samples, indicating that DEHP or its metabolites cross the placental barrier and result in exposure to the developing conceptus.204-206 DEHP levels in control feed were below the limit of detection (1.27 ppm) of the analytical method; however, detectable levels of MEHP were measured in control amniotic fluid and fetuses, but not in control dam plasma (GD 18). Detection of MEHP in control animal samples might have resulted from sample contamination during collection or analysis, due to the presence of phthalates in manufactured laboratory plasticware. Although not assessed in the present study, DEHP and MEHP can be transferred from dam to offspring via lactation.54 DEHP exposure during the perinatal period was associated with significantly decreased total and live litter size, due to a significantly decreased number of female pups per litter in the 10,000 ppm group (626 mg/kg/day). In previous studies, increased resorptions, postimplantation loss, and whole-litter loss have been observed following DEHP exposure at doses >500 mg/kg/day in pregnant rats.90,207-209 In a multigenerational reproductive assessment of DEHP, previously conducted by NTP, significant effects on litter size and sex ratio were observed following perinatal exposure of Sprague Dawley rat F0 dams at concentrations of 7,500 and 10,000 ppm.210 In this perinatal and postweaning study (Study 1), exposure-related decreases in birth and weaning mean body weights were observed in both male and female groups. Gestational DEHP exposure was associated with 15% and 12% decreases in postnatal day (PND) 1 mean body weights of 10,000 ppm male and female pups, respectively. Further growth retardation during lactation was observed with male and female pup weights. Postweaning, mean body weights of the 10,000 ppm offspring remained significantly decreased relative to control groups throughout the 2-year exposure period. The magnitude of effect on body weight observed in 10,000 ppm offspring was higher than the 30% decrease in postweaning body weights observed at the same dose level in the NTP multigeneration assessment of DEHP.210
No significant differences in overall survival were observed in either the perinatal and postweaning study (Study 1) or the postweaning-only study (Study 2) relative to concurrent control groups, although there was some early postweaning mortality (Study 1). Lower mean body weights (postweaning to study termination) were observed in 10,000 ppm male and female rats in both studies relative to control rats. In both studies, lower mean body weights were associated with lower body weight gain; however, the magnitude of effect was higher following perinatal and postweaning exposure compared to postweaning-only exposure, due to early life exposure that included gestation, lactation, and a brief period after weaning. In the 10,000 ppm male and female rat groups, the largest difference in feed consumption relative to the control groups occurred directly following weaning. In rats, increased rates of feed and water consumption, relative to body weight, are commonly observed in younger animals and decrease with subsequent growth and development. Furthermore, in the perinatal and postweaning study (Study 1), the 2-year direct exposure period began at weaning, 3 weeks earlier than in the postweaning-only study (PND 42) (Study 2). Although this 3-week interval represents a small fraction of the total exposure timeframe, perinatal and postweaning exposure groups were exposed to DEHP at earlier ages and therefore at higher doses than the corresponding groups in the postweaning-only study (Study 2), likely contributing to the 3%–20% higher mean chemical consumption (mg/kg/day) postweaning in Study 1 versus Study 2.
The following conclusions on the carcinogenicity of DEHP were determined based on the weight-of-evidence approach described in the Explanation of Levels of Evidence of Carcinogenic Activity. Conclusions on DEHP carcinogenic activity are described separately for the perinatal and postweaning and postweaning-only studies, based on the independent results observed in each study. Although some variability in carcinogenic outcomes was observed between rats exposed to DEHP during the perinatal and postweaning periods and those only exposed postweaning, it is unclear whether any differences correspond to specific developmental mechanisms during the perinatal period. Some noncarcinogenic outcomes were related to perinatal exposure, however, which included the gross lesions in the reproductive tract and histological lesions in the kidney.
At the conclusion of both studies, numerous neoplastic and nonneoplastic lesions in the liver were identified. In male rats, increased incidences of hepatocellular adenoma and hepatocellular adenoma or carcinoma (combined) were observed in both 2-year studies. In the perinatal and postweaning study (Study 1), there was an increase in rare hepatocellular carcinomas (historical control 2/489; range 0% to 2%) in the 10,000 ppm group (8.7%), whereas a positive trend was observed in these neoplasms in the postweaning-only study (Study 2). In both perinatal and postweaning and postweaning-only studies, higher incidences of hepatocellular cytoplasmic alteration, liver pigmentation, and liver necrosis were observed in male rats. Although considered minimal in severity, a higher incidence of hepatocellular hypertrophy was observed in 10,000 ppm male rats exposed during the perinatal and postweaning periods (35%) compared to male rats in the postweaning-only study (12%). Additionally, a significantly increased incidence of basophilic focus was observed in the livers of 10,000 ppm male rats in the perinatal and postweaning study, but not in their counterparts in the postweaning-only study (Study 2). Taken together, the significantly increased incidence of hepatocellular adenoma or carcinoma (combined) supported clear evidence of carcinogenicity in male rats in both 2-year studies.
In female rats, increased incidences of hepatocellular adenoma, hepatocellular carcinoma, and hepatocellular adenoma or carcinoma (combined) were observed in both 2-year studies. In female rats in the perinatal and postweaning study, the incidence of hepatocellular adenomas in the 3,000 ppm group that was higher (18%) than in the historical control range (15/489; range 0% to 8%); in females in the postweaning-only study (Study 2), the incidence of hepatocellular adenomas was above the historical control range in the 10,000 ppm group. Furthermore, an increased incidence of hepatocellular carcinoma (17%), a rare neoplasm type (historical control 1/489; range 0% to 2%), was also observed in 10,000 ppm female rats in the perinatal and postweaning study, whereas there was occurrence of hepatocellular carcinomas (0% versus 4% compared to the control group) in the 10,000 ppm group of the postweaning-only study (Study 2). Taken together, the increased incidence of hepatocellular adenoma or carcinoma (combined) supported clear evidence of carcinogenicity in the liver of female rats in both studies.
Significantly increased incidences of hepatocellular cytoplasmic alteration, hepatocellular hypertrophy, and liver pigmentation were observed in female rats in both 2-year studies. Higher incidences of eosinophilic foci were observed in 10,000 ppm female rats in the perinatal and postweaning study (Study 1), but not in the postweaning-only study (Study 2). A significantly increased incidence of hepatocellular carcinomas was observed only in 10,000 ppm females of the perinatal and postweaning study (17%) compared to the postweaning-only study (4%). This observation could be due to a higher rate of progression from hepatocellular adenoma to carcinoma because of early life or prolonged exposure, observations that are similar to those made in the perfluorooctanoic acid (PFOA) 2-year study211 in which perinatal and postweaning exposure led to a marginally higher carcinoma rate (4%) of this rare neoplasm in male rats relative to the rats with postweaning-only exposure (0%). Males with perinatal and postweaning exposure to DEHP also had a marginally higher hepatocellular carcinoma incidence compared to males with postweaning-only exposures.
Numerous chronic exposure studies have found that DEHP induces hepatic neoplasms in rats and mice. In the current study, estimated DEHP daily exposure concentrations (mg/kg/day) associated with higher incidences of hepatocellular adenomas or carcinomas were comparable to concentrations reported in previous studies. In a previous NTP study, chronic DEHP exposure via dosed feed resulted in increased incidences of hepatocellular carcinomas in male and female Fischer 344 (F344) rats at estimated daily exposures of 322 and 674 mg/kg/day in males and 394 and 774 mg/kg/day in females.69 In another study, increased incidences of hepatocellular adenomas and carcinomas were observed in male Sprague Dawley rats following lifetime exposure to 300 mg/kg/day.126 The precise mechanism by which DEHP induces hepatic neoplasms is not fully characterized. However, activation of peroxisome proliferator-activated receptor alpha (PPARα) by the DEHP proximal metabolite MEHP has been defined as a key molecular event by which DEHP causes hepatic neoplasms in rodents. The human relevance of mechanisms of carcinogenesis of the peroxisome proliferator class of chemicals is frequently debated.212 Additional research suggests that multiple signaling pathways and downstream mediators likely contribute to DEHP-induced hepatic carcinogenesis, rather than a single hallmark event such as activation of PPAR.73,212
Increased incidences of pancreatic acinar adenomas, carcinomas, and adenoma or carcinoma (combined) were observed in male rats in both studies. In both perinatal and postweaning and postweaning-only exposed male rats, increased incidences of pancreatic acinar adenomas occurred in the 3,000 and 10,000 ppm groups at higher rates than the historical control range (60/488; range 0% to 28%). Notably, a higher incidence of pancreatic cell adenomas occurred in 3,000 ppm perinatal and postweaning-exposed males (72%) when compared to males exposed to the same concentration postweaning-only (46%). Due to the potentially high background incidence of pancreatic acinar adenoma in the test rat strain (up to 28% in historical control groups), observed differences between perinatal and postweaning and postweaning-only exposure groups might have resulted from background variability of this lesion and not increased sensitivity related to perinatal exposure. A higher incidence of pancreatic acinar carcinoma, a rare neoplasm type (historical control 4/488; range 0% to 4%), was observed in 3,000 ppm male rats (6%) exposed during the perinatal and postweaning periods, and this neoplasm was increased in the 10,000 ppm male rats exposed postweaning-only. Furthermore, increased incidences of pancreatic acinar hyperplasia were noted in 3,000 and 10,000 ppm males exposed postweaning-only. The increased incidence of pancreatic acinar adenoma or carcinoma (combined) was considered clear evidence of carcinogenicity in male rats in both 2-year studies.
In female rats, occurrences of pancreatic acinar adenoma and carcinomas were observed in the postweaning-only study (Study 2), whereas occurrences of pancreatic acinar adenomas were observed in the perinatal and postweaning study (Study 1). In contrast to males, pancreatic acinar neoplasms are very rare in female rats (historic control 0/489). Occurrences of pancreatic acinus hyperplasia were also observed in exposed groups in both studies. After considering the rarity of this lesion type in female rats, the corroborating effect in male rats, and findings supportive of neoplastic progression, the incidence of pancreatic acinar adenoma or carcinoma (combined) was considered related to DEHP exposure in female rats.
Pancreatic acinar adenomas and carcinomas have been observed previously in male F344 rats following chronic DEHP exposure.86 Pancreatic adenomas have been reported in rodent models following exposure to various chemicals known to activate PPARα, such as PFOA, butyl benzyl phthalate (BBP), and Wyeth-14,643 (WY).211,213,214 Although direct activity of PPARα agonists on acinar cells has yet to be established, some data suggest that induction of pancreatic neoplasms by PPARα agonists is secondary to functional alterations in the liver. One proposed mode of action suggests that hepatic PPARα activation and subsequent alteration of transcriptional activity leads to alteration in bile acid composition and flow, resulting in cholestasis and increased expression of cholecystokinin (CCK).127 CCK is a growth factor reported to induce normal, adaptive, and neoplastic growth of pancreatic acinar cells in rats.215-217
Numerous gross lesions in the male reproductive tract were observed in male rats in the 10,000 ppm group exposed during both the perinatal and postweaning period, consistent with the “phthalate syndrome” suite of effects.99,218,219 These findings included decreased size of the phallus, testes, epididymides, prostate, seminal vesicles, and levator ani/bulbocavernosus (LABC) muscle; gubernacular length exceeding 20 mm; no gubernaculum present; nonregression of the cranial suspensory ligament (CSL); cleft phallus or prepuce; undescended testes (cryptorchid); epididymal agenesis (caput, corpus, or cauda); and incomplete preputial separation. All examined males exposed to 10,000 ppm DEHP in the perinatal and postweaning study presented with at least one of the aforementioned reproductive tract malformations; small or undescended testes were the most frequently observed reproductive tract malformations at 10,000 ppm. Male reproductive tract malformations have been observed in rodents following perinatal exposure to various phthalates, such as DEHP, di-n-butyl phthalate (DBP), di-isobutyl phthalate (DiBP), BBP, and diisononyl phthalate (DINP), and are indicative of hormone disruption of developmental androgen and insulin-like peptide 3 (Insl3) dependent signaling pathways.95,220,221 Differentiation of Wolffian structures (e.g., the epididymis, vas deferens, seminal vesicles) depends on fetal testosterone signaling, and masculinization of the prostate and external genitalia depends on the biosynthesis and signaling of the more potent androgen, dihydrotestosterone.222 Targeted disruption of Insl3 signaling alters gubernaculum development and CSL regression, leading to cryptorchidism.223,224 Developmental exposure to phthalates, such as DEHP, DBP, and BBP, disrupts Insl3 signaling leading to complete agenesis or hypoplasia of the gubernacular ligaments and retention of the testes in the inguinal or abdominal position.95,225 In the study presented here, undescended testes were consistently reduced in size and were more often retained in the abdominal region compared to the inguinal region.
Additional microscopic nonneoplastic lesions diagnosed in the testis and epididymis of male rats exposed during both the perinatal and postweaning periods were considered related to DEHP exposure. Observations of testicular germinal epithelium degeneration were noted in 10,000 ppm male rats with or without perinatal exposure and occurred concomitantly with epididymal hypospermia. Findings of testicular edema and exfoliated germ cells in the epididymal duct were observed in 10,000 ppm males of both studies; however, these effects were not significant in the perinatal and postweaning study and therefore considered related to exposure only in the postweaning-only study (Study 2). It is possible that evaluation at the end of a 2-year exposure led to lower sensitivity in detecting testicular degeneration as a higher incidence in control males can occur in older animals and findings are likely to be more evident in younger animals at lower exposure concentrations at earlier timepoints than in younger control animals.226,227 Seminiferous tubule dysgenesis was only present in 10,000 ppm males with perinatal and postweaning exposure (10/49). Seminiferous tubule dysgenesis is characterized as a developmental malformation seen microscopically as aberrant or misshapen seminiferous tubules, either with no or dilated lumens, which are often surrounded by focal Leydig cell aggregates. The Leydig cell aggregates within foci of dysgenesis differ morphologically from the Leydig cells in adenomas. The Leydig cells in these foci of dysgenesis appear to be poorly differentiated, are spindle-shaped and resemble embryonic Leydig cells, and do not have the abundant eosinophilic or vacuolated cytoplasm often apparent in hyperplasia or adenoma.226 Dysgenetic lesions might occur as one or more small foci per testis, and tend to be located near the center of the testis or might occupy the entire testis.226 The malformed tubules can appear to form anastomotic networks. The dysgenetic tubules contain poorly differentiated Sertoli cells, with small, elongated, and sometimes cleaved nuclei and less prominent nucleoli than the typical, prominent, tripartite nucleoli seen in mature Sertoli cells. Spermatogenesis is absent in these foci of dysgenesis but can be present elsewhere in the testis. Dysgenetic foci might be present in one or both testes and can be more severe in undescended than in scrotal testes.228,229 In men, similar microscopic dysgenetic foci have been reported in cryptorchid (undescended) testes,230 in testes also containing testicular cancer (both scrotal and cryptorchid testes231), and from testicular biopsies from the contralateral testis in men undergoing orchiectomy for testicular cancer.232
Increased pituitary pars distalis hypertrophy occurred in 10,000 ppm male rats in both studies. This lesion is commonly associated with disruption of the hypothalamus-pituitary-gonad signaling axis. Loss of negative feedback signaling by testicular-derived androgens, due to the antiandrogenic activity of phthalates, leads to increased hypothalamic release of gonadotropin-releasing hormone and subsequent increased releases of luteinizing hormone and follicle-stimulating hormone by gonadotrophs, or “castration cells,” in the pars distalis of the pituitary gland.233
There was a significant positive trend with testicular interstitial cell adenoma neoplasms in the postweaning-only study (Study 2), and the incidence of testicular interstitial cell adenoma (15/50; 30%) observed in 10,000 ppm male rats was above NTP’s historical control range (19/487; range 0% to 14%). However, there were no significant pairwise differences among the exposed groups compared to the control groups in the incidences of neoplasms or hyperplasias. Taken together, the data suggest that testicular interstitial cell adenomas may have been related to DEHP exposure in postweaning-only exposed male rats.
In contrast, perinatal and postweaning exposure did not increase the incidence of Leydig cell (interstitial) neoplasms relative to control animals, although the incidence of interstitial cell hyperplasia was considerably higher. Currently, it is unclear whether developmental malformations in the male reproductive tract, such as altered seminiferous tubule morphology or structural and functional alterations in Sertoli and Leydig cell populations, affect the carcinogenic potential of DEHP in testes in perinatally exposed rats relative to functional effects observed following adult exposure only. Increased incidences of focal interstitial cell hyperplasia were observed in both 2-year studies. Focal hyperplasia is considered a preneoplastic lesion that commonly forms as a part of a continuum leading to interstitial cell adenoma; it is distinct from diffuse hyperplasia, generally considered a physiological response to hormonal imbalance.234-236
Several PPARα agonists, including DEHP, PFOA, and WY, have been shown to induce Leydig cell neoplasms in rats.126,214 Multiple mechanisms by which PPARα agonists might induce testicular neoplasms through disruption of the hypothalamus-pituitary-thyroid axis have been postulated; however, the weight of evidence is currently inadequate to establish a mode of action.127 The marginal to no response in Leydig cell neoplasms to DEHP in this study is inconsistent with published studies and could be due to differential diagnoses. Varying morphological criteria distinguishing Leydig cell adenomas from seminiferous tubule dysgenesis may account for differential diagnoses226; the rodent strain studies may also be a factor as there were no Leydig cell neoplasms observed in the NTP PFOA studies,211 which used the Hsd:Sprague Dawley SD rat.
In female rats, increased incidences of adenoma, adenocarcinoma, squamous cell carcinoma, or squamous cell papilloma (combined) (mostly adenocarcinoma) were observed in the uterus (including cervix). Higher incidences of uterine (including cervix) adenoma, adenocarcinoma, squamous cell carcinoma, or squamous cell papilloma (combined) (26%) were observed in 10,000 ppm postweaning-only exposed females, which was above the NTP historical control incidence of this combination of lesions of 2% to 10%. Uterine inflammation was increased in all DEHP-exposed groups in the postweaning-only study (Study 2). This supported clear evidence of carcinogenic activity from DEHP postweaning-only exposure in female rats.
In the perinatal and postweaning study (Study 1), however, there was a marginally higher incidence of uterine neoplasms in DEHP-exposed groups compared to the control group and none of the pairwise comparisons to the control group were significant. The magnitude of the difference between the 10,000 ppm group and the control group in each study was lower in the perinatal and postweaning study (8% difference) compared to the postweaning-only study (22% difference). There was reduced certainty in this marginal response such that the incidence of uterine neoplasms may have been related to perinatal and postweaning exposure. The reason for this is not clear, but it is noteworthy that the testis and uterus, sites of endocrine action, had a lower response in general with the perinatal and postweaning exposure compared to the postweaning-only exposure.
The present study is the first to identify an association between DEHP exposure and induction of uterine neoplasms in female rats. The mechanism for this response is unclear. For previous chronic studies, no alterations in female reproductive organ histology were reported.69,86,123 Induction of the “tumor triad,” including liver, Leydig cell, and pancreatic acinar cell tumors, is a finding characteristic of sustained exposure to PPARα agonists in rats214; however, the relevance of this finding to humans is uncertain. The uterine neoplasm response in the NTP PFOA carcinogenicity study211 was considered equivocal evidence of carcinogenic activity. The magnitude of the response and level of evidence for PFOA is similar to that of the DEHP perinatal and postweaning study, whereas the response in the DEHP postweaning-only study was considerably higher. Further work will be required to assess the mode of action for these outcomes.
Evidence of DEHP-associated renal toxicity was specific to male and female rats with perinatal and postweaning exposure. Numerous nonneoplastic kidney lesions were increased in DEHP-exposed groups relative to control groups, such as papilla edema, papilla epithelium hyperplasia, papilla hemorrhage, infarct, and renal tubule cysts. Papillary edema was the most prevalent kidney lesion in 10,000 ppm male (39/49) and female (38/48) rats in the perinatal and postweaning study and was not present in the postweaning-only study (Study 2). This highly unusual bilateral lesion was characterized by marked dilation and/or distortion of the collecting ducts and moderate to marked expansion of the papilla interstitium by pale eosinophilic to fibrillary amphophilic material, consistent with edema. Periodic acid-Schiff (PAS) staining demonstrated that the basement of vascular structures, in addition to the renal tubule basement membranes in the cortex and medulla, were intact. An abrupt loss of PAS staining of the basement membranes of collecting tubules was observed, however, at the junction of the outer and inner medulla. Therefore, perinatal exposure to DEHP is presumed to interfere directly or indirectly with the proper development of the collecting tubules. The normal function of the collecting duct system is urine transport as well as electrolyte and fluid balance through reabsorption and excretion, processes regulated by aldosterone and vasopressin. Additional studies have reported DEHP-associated renal toxicity. Chronic dietary exposure to DEHP (≥789 mg/kg/day) was associated with increased severity of routinely occurring renal tubule pigmentation and chronic progressive nephropathy in male and female rats.86 Altered kidney function and kidney lesions have been reported in rats following developmental DEHP exposure. Impaired kidney development and function were observed in adult Wistar rats following maternal exposure to DEHP at doses of 0.25 and 6.25 mg/kg/day from GD 0 through offspring PND 21.237 Maternal exposure resulted in a decreased number of nephrons, higher glomerular volume, and smaller Bowman's capsule in offspring at weaning, as well as glomerulosclerosis, interstitial fibrosis, and effacement of podocyte foot processes in 33-week-old F1 rats. Taken together, these data suggest the developing kidney may be a sensitive target of DEHP toxicity.
Cardiovascular findings of increased heart valve fibrosis and thrombus were present in 10,000 ppm male rats in both of the present studies. Thrombosis in male rats has been associated with pancreatic cancer related to onset of an intrinsic hypercoagulable state caused by elevated activation of platelets and increased expression of procoagulant factors.238 However, a low concurrence of pancreatic cancer and cardiovascular thrombosis was observed in the present studies. Additionally, increased systolic blood pressure has been observed in rats and mice exposed to DEHP; however, this effect is thought to be secondary to renal dysfunction or alterations in renin and angiotensin II signaling.237,239
NTP has tested DEHP in a range of in vitro and in vivo genotoxicity assays, and the results were generally negative. The positive results seen in some of the in vitro assays for induction of sister chromatid exchanges (SCE) were seen in the presence of excessive cytotoxicity. The Organization for Economic Co-operation and Development test guideline240 for the in vitro SCE test was withdrawn in 2014, and the test is no longer requested by regulatory agencies. In vivo, the nonnegative responses that were observed in some of the NTP micronucleus assays were generally weak. The consensus from published data is that DEHP shows limited evidence of genotoxic potential, and for the sporadic positive results that have been reported, the response is either weak, not reproducible, obtained in a nonstandard test system, or qualified to some degree by the authors.
Lastly, carcinogenic responses that were related or may have been related to DEHP exposure were modeled to estimate benchmark doses corresponding to a 10% increase in neoplasm incidence (BMD10). For the similar target sites, the BMD10 levels based on the hepatocellular response were lower in males in the perinatal and postweaning study compared to the postweaning-only study (383 mg/kg/day and 434 mg/kg/day, respectively) and in females (123 mg/kg/day and 384 mg/kg/day, respectively). Conversely, BMD10 levels were lower for the pancreatic acinar neoplasm response in males with postweaning-only exposure compared with perinatal and postweaning exposure (31 mg/kg/day versus 86 mg/kg/day, respectively). The BMD10 for the neoplastic responses in the uterus in females was lower in the postweaning-only study compared to the perinatal and postweaning study (324 mg/kg/day versus 594 mg/kg/day, respectively). The lowest BMD10 levels were associated with incidences of pancreatic acinar adenoma or carcinoma (combined) in male rats in both studies. These data show no obvious overall increased sensitivity in carcinogenic response with perinatal and postweaning exposure compared to postweaning-only exposure, which is consistent with NTP’s study of PFOA perinatal exposure.211
References
1. National Center for Biotechnology Information (NCBI). PubChem Compound Summary: Bis(2-ethylhexyl) phthalate, CID 8343. Bethesda, MD: National Center for Biotechnology Information. https://pubchem.ncbi.nlm.nih.gov/compound/8343
2. National Institute for Occupational Safety and Health (NIOSH). NIOSH pocket guide to chemical hazards, di-sec octyl phthalate. U.S. Department of Health and Human Services, Centers for Disease Control and Prevention, National Institute for Occupational Safety and Health; 2019. https://www.cdc.gov/niosh/npg/npgd0236.html
3. U.S. Environmental Protection Agency (USEPA). CompTox Chemistry Dashboard: Di(2-ethylhexyl) phthalate 117-81-7 | DTXSID5020607. https://comptox.epa.gov/dashboard/DTXSID5020607
4. Agency for Toxic Substances and Disease Registry (ATSDR). Toxicological profile for di(2-ethylhexyl)phthalate (DEHP). Atlanta, GA: U.S. Department of Health and Human Services, Public Health Service; 2019.
5. U.S. Environmental Protection Agency (USEPA). Chemical data reporting (2012 and 2016 public CDR database). Washington, DC: U.S. Environmental Protection Agency, Office of Pollution Prevention and Toxics; 2017.
6. Toxics Use Reduction Institute (TURI). Five chemicals alternatives reduction study. Final report. Chapter 7. DEHP. Lowell, MA: The Massachusetts Toxics Use Reduction Institute, University of Massachusetts Lowell; 2005. https://www.turi.org/TURI_Publications/TURI_Methods_Policy_Reports/Five_Chemicals_Alternatives_Assessment_Study._2006
7. European Commission (EC). Bis(2-ethylhexyl)phthalate (DEHP) – Summary risk assessment report. Ispra (VA), Italy: European Chemicals Bureau, Institute for Health and Consumer Protection, Toxicology and Chemical Substance (TCS); 2008. EUR 23384EN/2.
8. Latini G, Ferri M, Chiellini F. Materials degradation in PVC medical devices, DEHP leaching and neonatal outcomes. Curr Med Chem. 2010; 17(26):2979-2989. DOI: 10.2174/092986710792064992 PubMed: 20858177
9. Bouattour Y, Wasiak M, Bernard L, Pinguet J, Richard D, Le Rouzo-Grèves M, Dhifallah I, Lambert C, Pereira B, Chennell P, et al. Quantification of bis(2-ethylhexyl) phthalate released by medical devices during respiratory assistance and estimation of patient exposure. Chemosphere. 2020; 255:126978. DOI: 10.1016/j.chemosphere.2020.126978 PubMed: 32417514
10. International Programme on Chemical Safety (IPCS). Environmental Health Criteria 131: Diethylhexyl phthalate. 1992.
11. Serrano SE, Braun J, Trasande L, Dills R, Sathyanarayana S. Phthalates and diet: A review of the food monitoring and epidemiology data. Environ Health. 2014; 13(1):43. DOI: 10.1186/1476-069X-13-43 PubMed: 24894065
12. Clark K, Cousins IT, Mackay D. Assessment of critical exposure pathways. In: Staples CA, editor. The Handbook of Environmental Chemistry. New York: Springer-Verlag; 2003. p. 227-262.
13. National Toxicology Program (NTP). NTP-CERHR monograph on the potential human reproductive and developmental effects of di (2-ethylhexyl) phthalate (DEHP). Research Triangle Park, NC: U.S. Department of Health and Human Services, National Institute of Environmental Health Sciences, National Toxicology Program; 2006. PubMed: 19407857
14. Fromme H, Lahrz T, Piloty M, Gebhart H, Oddoy A, Ruden H. Occurrence of phthalates and musk fragrances in indoor air and dust from apartments and kindergartens in Berlin (Germany). Indoor Air. 2004; 14(3):188-195. DOI: 10.1111/j.1600-0668.2004.00223.x PubMed: 15104786
15. Centers for Disease Control and Prevention (CDC). Fourth national report on human exposure to environmental chemicals. Updated tables, January 2019, volume one. Atlanta, GA: U.S. Department of Health and Human Services, Centers for Disease Control and Prevention; 2019. https://stacks.cdc.gov/view/cdc/75822
16. Wambaugh JF, Wang A, Dionisio KL, Frame A, Egeghy P, Judson R, Setzer RW. High throughput heuristics for prioritizing human exposure to environmental chemicals. Environ Sci Technol. 2014; 48(21):12760-12767. DOI: 10.1021/es503583j PubMed: 25343693
17. Dirven HA, van den Broek PH, Arends AM, Nordkamp HH, de Lepper AJ, Henderson PT, Jongeneelen FJ. Metabolites of the plasticizer di(2-ethylhexyl)phthalate in urine samples of workers in polyvinylchloride processing industries. Int Arch Occup Environ Health. 1993; 64(8):549-554. DOI: 10.1007/BF00517699 PubMed: 8314612
18. Nielsen J, Akesson B, Skerfving S. Phthalate ester exposure—air levels and health of workers processing polyvinylchloride. Am Ind Hyg Assoc J. 1985; 46(11):643-647. DOI: 10.1080/15298668591395463 PubMed: 4072908
19. Hines CJ, Hopf NBN, Deddens JA, Silva MJ, Calafat AM. Estimated daily intake of phthalates in occupationally exposed groups. J Expo Sci Environ Epidemiol. 2011; 21(2):133-141. DOI: 10.1038/jes.2009.62 PubMed: 20010977
20. Hines CJ, Nilsen Hopf NB, Deddens JA, Calafat AM, Silva MJ, Grote AA, Sammons DL. Urinary phthalate metabolite concentrations among workers in selected industries: A pilot biomonitoring study. Ann Occup Hyg. 2009; 53(1):1-17. DOI: 10.1093/annhyg/men066 PubMed: 18948546
21. Fong JP, Lee FJ, Lu IS, Uang SN, Lee CC. Estimating the contribution of inhalation exposure to di-2-ethylhexyl phthalate (DEHP) for PVC production workers, using personal air sampling and urinary metabolite monitoring. Int J Hyg Environ Health. 2014; 217(1):102-109. DOI: 10.1016/j.ijheh.2013.04.002 PubMed: 23665067
22. Wang W, Xu X, Fan CQ. Health hazard assessment of occupationally di-(2-ethylhexyl)-phthalate-exposed workers in China. Chemosphere. 2015; 120:37-44. DOI: 10.1016/j.chemosphere.2014.05.053 PubMed: 24974312
23. Ginsberg G, Ginsberg J, Foos B. Approaches to children’s exposure assessment: Case study with diethylhexylphthalate (DEHP). Int J Environ Res Public Health. 2016; 13(7). DOI: 10.3390/ijerph13070670 PubMed: 27376320
24. Zhu J, Phillips SP, Feng YL, Yang X. Phthalate esters in human milk: Concentration variations over a 6-month postpartum time. Environ Sci Technol. 2006; 40(17):5276-5281. DOI: 10.1021/es060356w PubMed: 16999099
25. Swedish National Chemicals Inspectorate (KEMI). Risk Assessment - bis(2-ethylhexyl) phthalate (CAS-No.: 117-81-7). 2000.
26. Silva MJ, Reidy JA, Herbert AR, Preau JL Jr, Needham LL, Calafat AM. Detection of phthalate metabolites in human amniotic fluid. Bull Environ Contam Toxicol. 2004; 72(6):1226-1231. DOI: 10.1007/s00128-004-0374-4 PubMed: 15362453
27. U.S. Food and Drug Administration (FDA). Safety assessment of di(2-ethylhexyl)phthalate (DEHP) released from PVC medical devices. Rockville, MD; 2001.
28. Den Braver-Sewradj SP, Piersma A, Hessel EVS. An update on the hazard of and exposure to diethyl hexyl phthalate (DEHP) alternatives used in medical devices. Crit Rev Toxicol. 2020; 50(8):650-672. DOI: 10.1080/10408444.2020.1816896 PubMed: 33006299
29. Tickner JA, Schettler T, Guidotti T, McCally M, Rossi M. Health risks posed by use of Di-2-ethylhexyl phthalate (DEHP) in PVC medical devices: A critical review. Am J Ind Med. 2001; 39(1):100-111. DOI: 10.1002/1097-0274(200101)39:1<100::aid-ajim10>3.0.co;2-q
30. 110th U.S. Congress. Consumer Product Safety Improvement Act of 2008. 2008.
31. Code of Federal Regulations (CFR). 2017; 17(Part 1307).
32. Code of Federal Regulations (CFR). 2001; 21(Part 177, Subpart B, Section 177.1010).
33. Code of Federal Regulations (CFR). 2001; 21(Part 177, Subpart B, Section 177.1200).
34. Carpenter CP, Weil CS, Smyth HF Jr. Chronic oral toxicity of di-(2-ethylhexyl) phthalate of rats, guinea pigs, and dogs. AMA Arch Ind Hyg Occup Med. 1953; 8(3):219-226. PubMed: 13079323
35. U.S. Environmental Protection Agency (USEPA). Integrated Risk Information System (IRIS), chemical assessment summary, di(2-ethylhexyl)phthalate; CASRN 117-81-7. Washington D.C.: U.S. Environmental Protection Agency, National Center for Environmental Assessment; 1987. https://cfpub.epa.gov/ncea/iris/iris_documents/documents/subst/0014_summary.pdf#nameddest=rfd
36. U.S. Environmental Protection Agency (USEPA). National Primary Drinking Water Regulations: Phthalate, di(2-Ethylhexyl). Washington, D.C.: Office of Water; 1995.
37. Occupational Safety and Health Administration (OSHA). Recommendations for consideration by the U.S. Secretary of Labor on the Adoption and use of occupational exposure limits by federal agencies. Occupational Safety and Health Administration, Federal Advisory Council on Occupational Safety and Health; 2012. https://www.osha.gov/dep/facosh/Exhibit_9b.pdf
38. Lake BG, Gray TJ, Foster JR, Stubberfield CR, Gangolli SD. Comparative studies on di-(2-ethylhexyl) phthalate-induced hepatic peroxisome proliferation in the rat and hamster. Toxicol Appl Pharmacol. 1984; 72(1):46-60. DOI: 10.1016/0041-008X(84)90248-5 PubMed: 6710484
39. Albro PW. Absorption, metabolism, and excretion of di(2-ethylhexyl) phthalate by rats and mice. Environ Health Perspect. 1986; 65:293-298. DOI: 10.1289/ehp.8665293 PubMed: 3086077
40. Albro PW, Corbett JT, Schroeder JL, Jordan S, Matthews HB. Pharmacokinetics, interactions with macromolecules and species differences in metabolism of DEHP. Environ Health Perspect. 1982; 45:19-25. DOI: 10.1289/ehp.824519 PubMed: 7140694
41. Rhodes C, Orton TC, Pratt IS, Batten PL, Bratt H, Jackson SJ, Elcombe CR. Comparative pharmacokinetics and subacute toxicity of di(2-ethylhexyl) phthalate (DEHP) in rats and marmosets: Extrapolation of effects in rodents to man. Environ Health Perspect. 1986; 65:299-307. DOI: 10.1289/ehp.8665299 PubMed: 3086078
42. Pollack GM, Li RC, Ermer JC, Shen DD. Effects of route of administration and repetitive dosing on the disposition kinetics of di(2-ethylhexyl) phthalate and its mono-de-esterified metabolite in rats. Toxicol Appl Pharmacol. 1985; 79(2):246-256. DOI: 10.1016/0041-008X(85)90346-1 PubMed: 4002226
43. Daniel JW, Bratt H. The absorption, metabolism and tissue distribution of di(2-ethylhexyl)phthalate in rats. Toxicology. 1974; 2(1):51-65. DOI: 10.1016/0300-483X(74)90042-0 PubMed: 4823738
44. Elsisi AE, Carter DE, Sipes IG. Dermal absorption of phthalate diesters in rats. Fundam Appl Toxicol. 1989; 12(1):70-77. DOI: 10.1016/0272-0590(89)90063-8 PubMed: 2925020
45. Tanaka A, Adachi T, Takahashi T, Yamaha T. Biochemical studies on phthalic esters I. Elimination, distribution and metabolism of di-(2-ethylhexyl)phthalate in rats. Toxicology. 1975; 4(2):253-264. DOI: 10.1016/0300-483X(75)90105-5 PubMed: 1154425
46. Ikeda GJ, Sapienza PP, Couvillion JL, Farber TM, van Loon EJ. Comparative distribution, excretion and metabolism of di-(2-ethylhexyl) phthalate in rats, dogs and miniature pigs. Food Cosmet Toxicol. 1980; 18(6):637-642. DOI: 10.1016/S0015-6264(80)80012-5 PubMed: 7203310
47. Chu I, Villeneuve DC, Secours V, Franklin C, Rock G, Viau A. Metabolism and tissue distribution of mono-2-ethylhexyl phthalate in the rat. Drug Metab Dispos. 1978; 6(2):146-149. PubMed: 26529
48. Lindgren A, Lindquist NG, Lyden A, Olsson T, Ullberg S. A whole body autoradiographic study on the distribution of 14C-labelled di-(2-ethylhexyl)phthalate in mice. Toxicology. 1982; 23(2-3):149-158. DOI: 10.1016/0300-483X(82)90094-4 PubMed: 6810506
49. Oishi S. Effects of phthalic acid esters on testicular mitochondrial functions in the rat. Arch Toxicol. 1990; 64(2):143-147. DOI: 10.1007/BF01974400 PubMed: 2350233
50. Sjöberg P, Bondesson U, Kjellen L, Lindquist NG, Montin G, Ploen L. Kinetics of di-(2-ethylhexyl) phthalate in immature and mature rats and effect on testis. Acta Pharmacol Toxicol (Copenh). 1985; 56(1):30-37. DOI: 10.1111/j.1600-0773.1985.tb01249.x PubMed: 3976401
51. Teirlynck OA, Belpaire F. Disposition of orally administered di-(2-ethylhexyl) phthalate and mono-(2-ethylhexyl) phthalate in the rat. Arch Toxicol. 1985; 57(4):226-230. DOI: 10.1007/BF00324782 PubMed: 4091646
52. Singh AR, Lawrence WH, Autian J. Maternal-fetal transfer of 14C-di-2-ethylhexyl phthalate and 14C-diethyl phthalate in rats. J Pharm Sci. 1975; 64(8):1347-1350. DOI: 10.1002/jps.2600640819 PubMed: 1151708
53. Srivastava S, Awasthi VK, Srivastava SP, Seth PK. Biochemical alterations in rat fetal liver following in utero exposure to di(2-ethylhexyl)phthalate (DEHP). Indian J Exp Biol. 1989; 27(10):885-888. PubMed: 2635147
54. Dostal LA, Weaver RP, Schwetz BA. Transfer of di(2-ethylhexyl) phthalate through rat milk and effects on milk composition and the mammary gland. Toxicol Appl Pharmacol. 1987; 91(3):315-325. DOI: 10.1016/0041-008X(87)90054-8 PubMed: 2892284
55. Sjöberg P, Lindqvist NG, Ploen L. Age-dependent response of the rat testes to di(2-ethylhexyl) phthalate. Environ Health Perspect. 1986; 65:237-242. DOI: 10.1289/ehp.65-1474698 PubMed: 3709447
56. Egestad B, Green G, Sjöberg P, Klasson-Wehler E, Gustafsson J. Chromatographic fractionation and analysis by mass spectrometry of conjugated metabolites of bis(2-ethylhexyl)phthalate in urine. J Chromatogr B Biomed Appl. 1996; 677(1):99-109. DOI: 10.1016/0378-4347(95)00439-4 PubMed: 8925107
57. Egestad B, Sjöberg P. Glucosidation as a new conjugation pathway for metabolites of bis(2-ethylhexyl) phthalate. Drug Metab Dispos. 1992; 20(3):470-472. PubMed: 1355727
58. Peck CC, Albro PW. Toxic potential of the plasticizer di(2-ethylhexyl) phthalate in the context of its disposition and metabolism in primates and man. Environ Health Perspect. 1982; 45:11-17. DOI: 10.1289/ehp.824511 PubMed: 7140682
59. Koch HM, Bolt HM, Di Angerer J. (2-ethylhexyl)phthalate (DEHP) metabolites in human urine and serum after a single oral dose of deuterium-labelled DEHP. Arch Toxicol. 2004; 78(3):123-130. DOI: 10.1007/s00204-003-0522-3 PubMed: 14576974
60. Koch HM, Bolt HM, Preuss R, Angerer J. New metabolites of di(2-ethylhexyl)phthalate (DEHP) in human urine and serum after single oral doses of deuterium-labelled DEHP. Arch Toxicol. 2005; 79(7):367-376. DOI: 10.1007/s00204-004-0642-4 PubMed: 15700144
61. Silva MJ, Samandar E, Preau JL Jr, Needham LL, Calafat AM. Urinary oxidative metabolites of di(2-ethylhexyl) phthalate in humans. Toxicology. 2006; 219(1-3):22-32. DOI: 10.1016/j.tox.2005.10.018 PubMed: 16332407
62. Kato K, Silva MJ, Reidy JA, Hurtz D 3rd, Malek NA, Needham LL, Nakazawa H, Barr DB, Calafat AM. Mono(2-ethyl-5-hydroxyhexyl) phthalate and mono-(2-ethyl-5-oxohexyl) phthalate as biomarkers for human exposure assessment to di-(2-ethylhexyl) phthalate. Environ Health Perspect. 2004; 112(3):327-330. DOI: 10.1289/ehp.6663 PubMed: 14998748
63. U.S. Consumer Product Safety Commission (CPSC). Toxicity review of DEHP. 2010. https://www.cpsc.gov/s3fs-public/ToxicityReviewOfDEHP.pdf
64. Shaffer CB, Carpenter CP, Smyth HF. Acute and subacute toxicity of di(2-ethylhexyl) phthalate with note upon its metabolism. J Ind Hyg Toxicol. 1945; 27(5):130-135.
65. Shibko SI, Blumenthal H. Toxicology of phthalic acid esters used in food-packaging material. Environ Health Perspect. 1973; 3:131-137. DOI: 10.1289/ehp.7303131 PubMed: 4704561
66. Lawrence WH, Malik M, Turner JE, Singh AR, Autian J. A toxicological investigation of some acute, short-term, and chronic effects of administering di-2-ethylhexyl phthalate (DEHP) and other phthalate esters. Environ Res. 1975; 9(1):1-11. DOI: 10.1016/0013-9351(75)90043-2 PubMed: 1122902
67. Krauskopf LG. Studies on the toxicity of phthalates via ingestion. Environ Health Perspect. 1973; 3:61-72. DOI: 10.1289/ehp.730361 PubMed: 4735798
68. Dostal LA, Jenkins WL, Schwetz BA. Hepatic peroxisome proliferation and hypolipidemic effects of di(2-ethylhexyl)phthalate in neonatal and adult rats. Toxicol Appl Pharmacol. 1987; 87(1):81-90. DOI: 10.1016/0041-008X(87)90086-X PubMed: 3798454
69. National Toxicology Program (NTP). NTP technical report on the carcinogenesis bioassay of di(2-ethylhexyl)phthalate (CAS No. 117-81-7) in F344 rats and B6C3F1 mice (feed study). Research Triangle Park, NC: U.S. Department of Health and Human Services, National Institute of Environmental Health Sciences, National Toxicology Program; 1982. NTP Technical Report No. 217. https://ntp.niehs.nih.gov/publications/reports/tr/200s/tr217/index.html?utm_source=direct&utm_medium=prod&utm_campaign=ntpgolinks&utm_term=tr217abs PubMed: 12778218
70. Crocker JF, Safe SH, Acott P. Effects of chronic phthalate exposure on the kidney. J Toxicol Environ Health. 1988; 23(4):433-444. DOI: 10.1080/15287398809531126 PubMed: 3361614
71. Moore MR. Oncogenicity study in rats with di(2-ethylhexyl)phthalate including ancillary hepatocellular proliferation and biochemical analyses. Corning Hazleton Incorporated (CHV); 1996.
72. Moore MR. Oncogenicity study in mice with Di (2-ethylhexyl)phthalate including ancillary hepatocellular proliferation and biochemical analyses. Corning Hazleton Incorporated (CHV); 1997.
73. International Agency for Research on Cancer (IARC). Evaluation of Carcinogenic Risks to Humans: Some Industrial Chemicals. Lyon, France; 2000.
74. Ward JM, Peters JM, Perella CM, Gonzalez FJ. Receptor and nonreceptor-mediated organ-specific toxicity of di(2-ethylhexyl)phthalate (DEHP) in peroxisome proliferator-activated receptor alpha-null mice. Toxicol Pathol. 1998; 26(2):240-246. DOI: 10.1177/019262339802600208 PubMed: 9547862
75. Maloney EK, Waxman DJ. Trans-activation of PPARalpha and PPARgamma by structurally diverse environmental chemicals. Toxicol Appl Pharmacol. 1999; 161(2):209-218. DOI: 10.1006/taap.1999.8809 PubMed: 10581215
76. Dostal LA, Chapin RE, Stefanski SA, Harris MW, Schwetz BA. Testicular toxicity and reduced Sertoli cell numbers in neonatal rats by di(2-ethylhexyl)phthalate and the recovery of fertility as adults. Toxicol Appl Pharmacol. 1988; 95(1):104-121. DOI: 10.1016/S0041-008X(88)80012-7 PubMed: 3413790
77. Gray TJ, Butterworth KR. Testicular atrophy produced by phthalate esters. Arch Toxicol Suppl. 1980; 4:452-455. DOI: 10.1007/978-3-642-67729-8_106 PubMed: 6776936
78. Gray TJ, Gangolli SD. Aspects of the testicular toxicity of phthalate esters. Environ Health Perspect. 1986; 65:229-235. DOI: 10.1289/ehp.8665229 PubMed: 3709446
79. Lamb JC, Chapin RE, Teague J, Lawton AD, Reel JR. Reproductive effects of four phthalic acid esters in the mouse. Toxicol Appl Pharmacol. 1987; 88(2):255-269. DOI: 10.1016/0041-008X(87)90011-1 PubMed: 3564043
80. Parmar D, Srivastava SP, Singh GB, Seth PK. Testicular toxicity of Di(2-ethylhexyl)phthalate in developing rats. Vet Hum Toxicol. 1995; 37(4):310-313. PubMed: 8540215
81. Poon R, Lecavalier P, Mueller R, Valli VE, Procter BG, Chu I. Subchronic oral toxicity of di-n-octyl phthalate and di(2-Ethylhexyl) phthalate in the rat. Food Chem Toxicol. 1997; 35(2):225-239. DOI: 10.1016/S0278-6915(96)00064-6 PubMed: 9146736
82. Sjöberg P, Bondesson U, Gray TJ, Ploen L. Effects of di-(2-ethylhexyl) phthalate and five of its metabolites on rat testis in vivo and in in vitro. Acta Pharmacol Toxicol (Copenh). 1986; 58(3):225-233. DOI: 10.1111/j.1600-0773.1986.tb00098.x PubMed: 3716815
83. Chapin RE, Gray TJ, Phelps JL, Dutton SL. The effects of mono-(2-ethylhexyl)-phthalate on rat Sertoli cell-enriched primary cultures. Toxicol Appl Pharmacol. 1988; 92(3):467-479. DOI: 10.1016/0041-008X(88)90186-X PubMed: 3353991
84. Svechnikov K, Svechnikova I, Söder O. Inhibitory effects of mono-ethylhexyl phthalate on steroidogenesis in immature and adult rat Leydig cells in vitro. Reprod Toxicol. 2008; 25(4):485-490. DOI: 10.1016/j.reprotox.2008.05.057 PubMed: 18583092
85. Parmar D, Srivastava SP, Singh GB, Seth PK. Effect of testosterone on the testicular atrophy caused by di(2-ethylhexyl)phthalate (DEHP). Toxicol Lett. 1987; 36(3):297-308. DOI: 10.1016/0378-4274(87)90199-8 PubMed: 2884757
86. David RM, Moore MR, Finney DC, Guest D. Chronic toxicity of di(2-ethylhexyl)phthalate in rats. Toxicol Sci. 2000; 55(2):433-443. DOI: 10.1093/toxsci/55.2.433 PubMed: 10828276
87. Pugh G Jr, Isenberg JS, Kamendulis LM, Ackley DC, Clare LJ, Brown R, Lington AW, Smith JH, Klaunig JE. Effects of di-isononyl phthalate, di-2-ethylhexyl phthalate, and clofibrate in cynomolgus monkeys. Toxicol Sci. 2000; 56(1):181-188. DOI: 10.1093/toxsci/56.1.181 PubMed: 10869467
88. Davis BJ, Maronpot RR, Heindel JJ. Di-(2-ethylhexyl) phthalate suppresses estradiol and ovulation in cycling rats. Toxicol Appl Pharmacol. 1994; 128(2):216-223. DOI: 10.1006/taap.1994.1200 PubMed: 7940536
89. Ritter EJ, Scott WJ Jr, Randall JL, Ritter JM. Teratogenicity of di(2-ethylhexyl) phthalate, 2-ethylhexanol, 2-ethylhexanoic acid, and valproic acid, and potentiation by caffeine. Teratology. 1987; 35(1):41-46. DOI: 10.1002/tera.1420350107 PubMed: 3105103
90. Tyl RW, Price CJ, Marr MC, Kimmel CA. Developmental toxicity evaluation of dietary di(2-ethylhexyl)phthalate in Fischer 344 rats and CD-1 mice. Fundam Appl Toxicol. 1988; 10(3):395-412. DOI: 10.1016/0272-0590(88)90286-2 PubMed: 3371580
91. George F, Wilson J. Gonads and ducts in mammals.In: The Physiology of Reproduction. 1994. p. 3-28.
92. Byskov AG, Hoyer PE. Embryology of mammalian gonads and ducts. In: Knobil E, Neill JD, editors. The Physiology of Reproduction. New York: Raven Press; 1994. p. 487-539.
93. Parks LG, Ostby JS, Lambright CR, Abbott BD, Klinefelter GR, Barlow NJ, Gray LE Jr. The plasticizer diethylhexyl phthalate induces malformations by decreasing fetal testosterone synthesis during sexual differentiation in the male rat. Toxicol Sci. 2000; 58(2):339-349. DOI: 10.1093/toxsci/58.2.339 PubMed: 11099646
94. Kay VR, Bloom MS, Foster WG. Reproductive and developmental effects of phthalate diesters in males. Crit Rev Toxicol. 2014; 44(6):467-498. DOI: 10.3109/10408444.2013.875983 PubMed: 24903855
95. Gray LE Jr, Ostby J, Furr J, Price M, Veeramachaneni DN, Parks L. Perinatal exposure to the phthalates DEHP, BBP, and DINP, but not DEP, DMP, or DOTP, alters sexual differentiation of the male rat. Toxicol Sci. 2000; 58(2):350-365. DOI: 10.1093/toxsci/58.2.350 PubMed: 11099647
96. Wolf C, Lambright C, Mann P, Price M, Cooper RL, Ostby J, Gray LE Jr. Administration of potentially antiandrogenic pesticides (procymidone, linuron, iprodione, chlozolinate, p, p′-DDE, and ketoconazole) and toxic substances (dibutyl-and diethylhexyl phthalate, PCB 169, and ethane dimethane sulphonate) during sexual differentiation produces diverse profiles of reproductive malformations in the male rat. Toxicol Ind Health. 1999; 15(1-2):94-118. DOI: 10.1177/074823379901500109 PubMed: 10188194
97. Foster PM, Mylchreest E, Gaido KW, Sar M. Effects of phthalate esters on the developing reproductive tract of male rats. Hum Reprod Update. 2001; 7(3):231-235. DOI: 10.1093/humupd/7.3.231 PubMed: 11392369
98. Arcadi FA, Costa C, Imperatore C, Marchese A, Rapisarda A, Salemi M, Trimarchi GR, Costa G. Oral toxicity of bis(2-ethylhexyl) phthalate during pregnancy and suckling in the Long-Evans rat. Food Chem Toxicol. 1998; 36(11):963-970. DOI: 10.1016/S0278-6915(98)00065-9 PubMed: 9771559
99. Foster PM. Disruption of reproductive development in male rat offspring following in utero exposure to phthalate esters. Int J Androl. 2006; 29(1):140-147, discussion 181-145. DOI: 10.1111/j.1365-2605.2005.00563.x PubMed: 16102138
100. National Toxicology Program (NTP). Diethylhexylphthalate: Multigenerational reproductive assessment by continuous breeding when administered to Sprague-Dawley rats in the diet. Research Triangle Park, NC: U.S. Department of Health and Human Services, National Institute of Environmental Health Sciences, National Toxicology Program; 2004.
101. Duty SM, Calafat AM, Silva MJ, Brock JW, Ryan L, Chen Z, Overstreet J, Hauser R. The relationship between environmental exposure to phthalates and computer-aided sperm analysis motion parameters. J Androl. 2004; 25(2):293-302. DOI: 10.1002/j.1939-4640.2004.tb02790.x PubMed: 14760016
102. Duty SM, Calafat AM, Silva MJ, Ryan L, Hauser R. Phthalate exposure and reproductive hormones in adult men. Hum Reprod. 2005; 20(3):604-610. DOI: 10.1093/humrep/deh656 PubMed: 15591081
103. Duty SM, Silva MJ, Barr DB, Brock JW, Ryan L, Chen Z, Herrick RF, Christiani DC, Hauser R. Phthalate exposure and human semen parameters. Epidemiology. 2003; 14(3):269-277. DOI: 10.1097/01.EDE.0000059950.11836.16 PubMed: 12859026
104. Duty SM, Singh NP, Silva MJ, Barr DB, Brock JW, Ryan L, Herrick RF, Christiani DC, Hauser R. The relationship between environmental exposures to phthalates and DNA damage in human sperm using the neutral comet assay. Environ Health Perspect. 2003; 111(9):1164-1169. DOI: 10.1289/ehp.5756 PubMed: 12842768
105. Hauser R, Williams P, Altshul L, Calafat AM. Evidence of interaction between polychlorinated biphenyls and phthalates in relation to human sperm motility. Environ Health Perspect. 2005; 113(4):425-430. DOI: 10.1289/ehp.7305 PubMed: 15811833
106. Swan SH. Environmental phthalate exposure in relation to reproductive outcomes and other health endpoints in humans. Environ Res. 2008; 108(2):177-184. DOI: 10.1016/j.envres.2008.08.007 PubMed: 18949837
107. Swan SH, Main KM, Liu F, Stewart SL, Kruse RL, Calafat AM, Mao CS, Redmon JB, Ternand CL, Sullivan S, et al. Decrease in anogenital distance among male infants with prenatal phthalate exposure. Environ Health Perspect. 2005; 113(8):1056-1061. DOI: 10.1289/ehp.8100 PubMed: 16079079
108. Sharpe RM, Skakkebaek NE. Testicular dysgenesis syndrome: Mechanistic insights and potential new downstream effects. Fertil Steril. 2008; 89(2 Suppl):e33-e38. DOI: 10.1016/j.fertnstert.2007.12.026 PubMed: 18308057
109. Skakkebæk NE, Rajpert-De Meyts E, Main KM. Testicular dysgenesis syndrome: An increasingly common developmental disorder with environmental aspects. Hum Reprod. 2001; 16(5):972-978. DOI: 10.1093/humrep/16.5.972 PubMed: 11331648
110. Larsen ST, Hansen JS, Thygesen P, Begtrup M, Poulsen OM, Nielsen GD. Adjuvant and immuno-suppressive effect of six monophthalates in a subcutaneous injection model with BALB/c mice. Toxicology. 2001; 169(1):37-51. DOI: 10.1016/S0300-483X(01)00484-X PubMed: 11696408
111. Badr MZ, Shnyra A, Zoubine M, Norkin M, Herndon B, Quinn T, Miranda RN, Cunningham ML, Molteni A. Phthalate-induced liver protection against deleterious effects of the Th1 response: A potentially serious health hazard. PPAR Res. 2007; 2007:49671. DOI: 10.1155/2007/49671 PubMed: 18566640
112. Larsen ST, Nielsen GD. The adjuvant effect of di-(2-ethylhexyl) phthalate is mediated through a PPARalpha-independent mechanism. Toxicol Lett. 2007; 170(3):223-228. DOI: 10.1016/j.toxlet.2007.03.009 PubMed: 17462839
113. Tonk EC, Verhoef A, Gremmer ER, van Loveren H, Piersma AH. Relative sensitivity of developmental and immune parameters in juvenile versus adult male rats after exposure to di(2-ethylhexyl) phthalate. Toxicol Appl Pharmacol. 2012; 260(1):48-57. DOI: 10.1016/j.taap.2012.01.018 PubMed: 22310177
114. Piepenbrink MS, Hussain I, Marsh JA, Dietert RR. Developmental immunotoxicology of di-(2-ethylhexyl)phthalate (DEHP): Age-based assessment in the female rat. J Immunotoxicol. 2005; 2(1):21-31. DOI: 10.1080/15363750490429435 PubMed: 18958656
115. Butala JH, David RM, Gans G, McKee RH, Guo TL, Peachee VL, White KL Jr. Phthalate treatment does not influence levels of IgE or Th2 cytokines in B6C3F1 mice. Toxicology. 2004; 201(1-3):77-85. DOI: 10.1016/j.tox.2004.04.004 PubMed: 15297022
116. Hansen JS, Larsen ST, Poulsen LK, Nielsen GD. Adjuvant effects of inhaled mono-2-ethylhexyl phthalate in BALB/cJ mice. Toxicology. 2007; 232(1-2):79-88. DOI: 10.1016/j.tox.2006.12.011 PubMed: 17241728
117. Larsen ST, Hansen JS, Hammer M, Alarie Y, Nielsen GD. Effects of mono-2-ethylhexyl phthalate on the respiratory tract in BALB/c mice. Hum Exp Toxicol. 2004; 23(11):537-545. DOI: 10.1191/0960327104ht486oa PubMed: 15625780
118. Jaakkola JJ, Knight TL. The role of exposure to phthalates from polyvinyl chloride products in the development of asthma and allergies: A systematic review and meta-analysis. Environ Health Perspect. 2008; 116(7):845-853. DOI: 10.1289/ehp.10846 PubMed: 18629304
119. Nielsen J, Fahraeus C, Bensryd I, Akesson B, Welinder H, Linden K, Skerfving S. Small airways function in workers processing polyvinylchloride. Int Arch Occup Environ Health. 1989; 61(7):427-430. DOI: 10.1007/BF00386474 PubMed: 2777385
120. Jaakkola JJ, Ieromnimon A, Jaakkola MS. Interior surface materials and asthma in adults: A population-based incident case-control study. Am J Epidemiol. 2006; 164(8):742-749. DOI: 10.1093/aje/kwj249 PubMed: 16877535
121. Bornehag CG, Sundell J, Weschler CJ, Sigsgaard T, Lundgren B, Hasselgren M, Hagerhed-Engman L. The association between asthma and allergic symptoms in children and phthalates in house dust: A nested case-control study. Environ Health Perspect. 2004; 112(14):1393-1397. DOI: 10.1289/ehp.7187 PubMed: 15471731
122. Kolarik B, Naydenov K, Larsson M, Bornehag CG, Sundell J. The association between phthalates in dust and allergic diseases among Bulgarian children. Environ Health Perspect. 2008; 116(1):98-103. DOI: 10.1289/ehp.10498 PubMed: 18197306
123. Kluwe WM, McConnell EE, Huff JE, Haseman JK, Douglas JF, Hartwell WV. Carcinogenicity testing of phthalate esters and related compounds by the National Toxicology Program and the National Cancer Institute. Environ Health Perspect. 1982; 45:129-133. DOI: 10.1289/ehp.8245129 PubMed: 7140685
124. David RM, Moore MR, Cifone MA, Finney DC, Guest D. Chronic peroxisome proliferation and hepatomegaly associated with the hepatocellular tumorigenesis of di(2-ethylhexyl)phthalate and the effects of recovery. Toxicol Sci. 1999; 50(2):195-205. DOI: 10.1093/toxsci/50.2.195 PubMed: 10478855
125. David RM, Moore MR, Finney DC, Guest D. Chronic toxicity of di(2-ethylhexyl)phthalate in mice. Toxicol Sci. 2000; 58(2):377-385. DOI: 10.1093/toxsci/58.2.377 PubMed: 11099649
126. Voss C, Zerban H, Bannasch P, Berger MR. Lifelong exposure to di-(2-ethylhexyl)-phthalate induces tumors in liver and testes of Sprague-Dawley rats. Toxicology. 2005; 206(3):359-371. DOI: 10.1016/j.tox.2004.07.016 PubMed: 15588926
127. Klaunig JE, Babich MA, Baetcke KP, Cook JC, Corton JC, David RM, DeLuca JG, Lai DY, McKee RH, Peters JM, et al. PPARalpha agonist-induced rodent tumors: Modes of action and human relevance. Crit Rev Toxicol. 2003; 33(6):655-780. DOI: 10.1080/713608372 PubMed: 14727734
128. National Toxicology Program (NTP). Report on Carcinogens, Fourteenth Edition. Research Triangle Park, NC: U.S. Department of Health and Human Services, National Institute of Environmental Health Sciences, National Toxicology Program; 2016. https://ntp.niehs.nih.gov/go/roc14
129. Grosse Y, Baan R, Secretan-Lauby B, El Ghissassi F, Bouvard V, Benbrahim-Tallaa L, Guha N, Islami F, Galichet L, Straif K, et al. Carcinogenicity of chemicals in industrial and consumer products, food contaminants and flavourings, and water chlorination byproducts. Lancet Oncol. 2011; 12(4):328-329. DOI: 10.1016/S1470-2045(11)70088-2 PubMed: 21598447
130. Ito Y, Yamanoshita O, Asaeda N, Tagawa Y, Lee CH, Aoyama T, Ichihara G, Furuhashi K, Kamijima M, Gonzalez FJ, et al. Di(2-ethylhexyl)phthalate induces hepatic tumorigenesis through a peroxisome proliferator-activated receptor alpha-independent pathway. J Occup Health. 2007; 49(3):172-182. DOI: 10.1539/joh.49.172 PubMed: 17575397
131. Morimura K, Cheung C, Ward JM, Reddy JK, Gonzalez FJ. Differential susceptibility of mice humanized for peroxisome proliferator-activated receptor alpha to Wy-14,643-induced liver tumorigenesis. Carcinogenesis. 2006; 27(5):1074-1080. DOI: 10.1093/carcin/bgi329 PubMed: 16377806
132. Yang Q, Ito S, Gonzalez FJ. Hepatocyte-restricted constitutive activation of PPAR alpha induces hepatoproliferation but not hepatocarcinogenesis. Carcinogenesis. 2007; 28(6):1171-1177. DOI: 10.1093/carcin/bgm046 PubMed: 17331954
133. López-Carrillo L, Hernández-Ramírez RU, Calafat AM, Torres-Sánchez L, Galván-Portillo M, Needham LL, Ruiz-Ramos R, Cebrián ME. Exposure to phthalates and breast cancer risk in northern Mexico. Environ Health Perspect. 2010; 118(4):539-544. DOI: 10.1289/ehp.0901091 PubMed: 20368132
134. Hagmar L, Akesson B, Nielsen J, Andersson C, Lindén K, Attewell R, Möller T. Mortality and cancer morbidity in workers exposed to low levels of vinyl chloride monomer at a polyvinyl chloride processing plant. Am J Ind Med. 1990; 17(5):553-565. DOI: 10.1002/ajim.4700170502 PubMed: 2337081
135. Hardell L, Ohlson CG, Fredrikson M. Occupational exposure to polyvinyl chloride as a risk factor for testicular cancer evaluated in a case-control study. Int J Cancer. 1997; 73(6):828-830. DOI: 10.1002/(sici)1097-0215(19971210)73:6<828::aid-ijc10>3.0.co;2-0
136. Ohlson CG, Hardell L. Testicular cancer and occupational exposures with a focus on xenoestrogens in polyvinyl chloride plastics. Chemosphere. 2000; 40(9-11):1277-1282. DOI: 10.1016/S0045-6535(99)00380-X PubMed: 10739073
137. Hansen J. Risk for testicular cancer after occupational exposure to plastics. Int J Cancer. 1999; 82(6):911-912. DOI: 10.1002/(sici)1097-0215(19990909)82:6%3c911::aid-ijc23%3e3.0.co;2-o
138. Hardell L, Malmqvist N, Ohlson CG, Westberg H, Eriksson M. Testicular cancer and occupational exposure to polyvinyl chloride plastics: A case-control study. Int J Cancer. 2004; 109(3):425-429. DOI: 10.1002/ijc.11709 PubMed: 14961582
139. Westberg HB, Hardell LO, Malmqvist N, Ohlson CG, Axelson O. On the use of different measures of exposure-experiences from a case-control study on testicular cancer and PVC exposure. J Occup Environ Hyg. 2005; 2(7):351-356. DOI: 10.1080/15459620590969046 PubMed: 16020098
140. Selenskas S, Teta MJ, Vitale JN. Pancreatic cancer among workers processing synthetic resins. Am J Ind Med. 1995; 28(3):385-398. DOI: 10.1002/ajim.4700280308 PubMed: 7485192
141. Huber WW, Grasl-Kraupp B, Schulte-Hermann R. Hepatocarcinogenic potential of di(2-ethylhexyl)phthalate in rodents and its implications on human risk. Crit Rev Toxicol. 1996; 26(4):365-481. DOI: 10.3109/10408449609048302 PubMed: 8817083
142. International Agency for Research on Cancer (IARC). IARC monographs on the evaluation of the carcinogenic risk of chemicals to humans. Some industrial chemicals and dyestuffs. Lyon, France; 1982.
143. Caldwell JC. DEHP: Genotoxicity and potential carcinogenic mechanisms—A review. Mutat Res. 2012; 751(2):82-157. DOI: 10.1016/j.mrrev.2012.03.001 PubMed: 22484601
144. Tomita I, Nakamura Y, Aoki N, Inui N. Mutagenic/carcinogenic potential of DEHP and MEHP. Environ Health Perspect. 1982; 45:119-125. DOI: 10.1289/ehp.8245119 PubMed: 6814903
145. Chang YJ, Tseng CY, Lin PY, Chuang YC, Chao MW. Acute exposure to DEHP metabolite, MEHP cause genotoxicity, mutagenesis and carcinogenicity in mammalian Chinese hamster ovary cells. Carcinogenesis. 2017; 38(3):336-345. DOI: 10.1093/carcin/bgx009 PubMed: 28426879
146. Erkekoglu P, Rachidi W, Yuzugullu OG, Giray B, Favier A, Ozturk M, Hincal F. Evaluation of cytotoxicity and oxidative DNA damaging effects of di(2-ethylhexyl)-phthalate (DEHP) and mono(2-ethylhexyl)-phthalate (MEHP) on MA-10 Leydig cells and protection by selenium. Toxicol Appl Pharmacol. 2010; 248(1):52-62. DOI: 10.1016/j.taap.2010.07.016 PubMed: 20659492
147. Erkekoğlu P, Rachidi W, De Rosa V, Giray B, Favier A, Hincal F. Protective effect of selenium supplementation on the genotoxicity of di(2-ethylhexyl)phthalate and mono(2-ethylhexyl)phthalate treatment in LNCaP cells. Free Radic Biol Med. 2010; 49(4):559-566. DOI: 10.1016/j.freeradbiomed.2010.04.038 PubMed: 20466057
148. Park SY, Choi J. Cytotoxicity, genotoxicity and ecotoxicity assay using human cell and environmental species for the screening of the risk from pollutant exposure. Environ Int. 2007; 33(6):817-822. DOI: 10.1016/j.envint.2007.03.014 PubMed: 17499852
149. Zeiger E, Haworth S, Mortelmans K, Speck W. Mutagenicity testing of di(2-ethylhexyl)phthalate and related chemicals in Salmonella. Environ Mutagen. 1985; 7(2):213-232. DOI: 10.1002/em.2860070209 PubMed: 3971959
150. Zeiger E, Haworth S. Tests with a preincubation modification of the Salmonella/microsome assay. In: Ashby J, de Serres FJ, editors. Progress in Mutation Research: Evaluation of short-term tests for carcinogens: Report of the International Programme on Chemical Safety’s Collaborative Study on In Vitro Assays. Elsevier; 1985. p. 187-199.
151. Myhr B, Bowers L, Caspary WJ. Assays for the induction of gene mutations at the thymidine kinase locus in L5178Y mouse lymphoma cells in culture. In: Ashby J, de Serres FJ, editors. Progress in Mutation Research: Evaluation of short-term tests for carcinogens: Report of the International Programme on Chemical Safety’s Collaborative Study on In Vitro Assays. Elsevier Science Publisher; 1985. p. 555-568.
152. Simon VF, Kauhanen K, Tardiff RG. Mutagenic activity of chemicals identified in drinking water. In: Scott D, Bridges BA, Sobels FH, editors. Progress in Genetic Toxicology: Proceedings of the Second International Conference on Environmental Mutagens. Elsevier; 1977. p. 249-258.
153. Gulati DK, Witt K, Anderson B, Zeiger E, Shelby MD. Chromosome aberration and sister chromatid exchange tests in Chinese hamster ovary cells in vitro III: Results with 27 chemicals. Environ Mol Mutagen. 1989; 13(2):133-193. DOI: 10.1002/em.2850130208 PubMed: 2917552
154. Stenchever MA, Allen MA, Jerominski L, Petersen RV. Effects of bis(2-ethylhexyl) phthalate on chromosomes of human leukocytes and human fetal lung cells. J Pharm Sci. 1976; 65(11):1648-1651. DOI: 10.1002/jps.2600651121 PubMed: 993999
155. Abe S, Sasaki M. Chromosome aberrations and sister chromatid exchanges in Chinese hamster cells exposed to various chemicals. J Natl Cancer Inst. 1977; 58(6):1635-1641. DOI: 10.1093/jnci/58.6.1635 PubMed: 864744
156. Ishidate M Jr, Odashima S. Chromosome tests with 134 compounds on Chinese hamster cells in vitro—a screening for chemical carcinogens. Mutat Res. 1977; 48(3-4):337-353. DOI: 10.1016/0027-5107(77)90177-4 PubMed: 876270
157. Phillips BJ, James TEB, Gangoli SD. Genotoxicity studies of di(2-ethylhexyl)phthalate and its metabolites in CHO cells. Mutat Res. 1982; 102(3):297-304. DOI: 10.1016/0165-1218(82)90139-2 PubMed: 6890626
158. Yoon JS, Mason JM, Valencia R, Woodruff RC, Zimmering S. Chemical mutagenesis testing in Drosophila. IV. Results of 45 coded compounds tested for the National Toxicology Program. Environ Mutagen. 1985; 7(3):349-367. DOI: 10.1002/em.2860070310 PubMed: 3930235
159. Zimmering S, Mason JM, Valencia R. Chemical mutagenesis testing in Drosophila. VII. Results of 22 coded compounds tested in larval feeding experiments. Environ Mol Mutagen. 1989; 14(4):245-251. DOI: 10.1002/em.2850140406 PubMed: 2583131
160. Kitamoto S, Matsuyama R, Uematsu Y, Ogata K, Ota M, Yamada T, Miyata K, Kimura J, Funabashi H, Saito K. Genotoxicity evaluation of benzene, di(2-ethylhexyl) phthalate, and trisodium ethylenediamine tetraacetic acid monohydrate using a combined rat comet/micronucleus assays. Mutat Res Genet Toxicol Environ Mutagen. 2015; 786-788:137-143. DOI: 10.1016/j.mrgentox.2015.05.002 PubMed: 26212304
161. Akagi J-I, Toyoda T, Cho Y-M, Mizuta Y, Nohmi T, Nishikawa A, Ogawa K. Validation study of the combined repeated-dose toxicity and genotoxicity assay using gpt delta rats. Cancer Sci. 2015; 106(5):529-541. DOI: 10.1111/cas.12634 PubMed: 25683344
162. Kanki K, Nishikawa A, Masumura K, Umemura T, Imazawa T, Kitamura Y, Nohmi T, Hirose M. In vivo mutational analysis of liver DNA in gpt delta transgenic rats treated with the hepatocarcinogens N-nitrosopyrrolidine, 2-amino-3-methylimidazo[4,5-f]quinoline, and di(2-ethylhexyl)phthalate. Mol Carcinog. 2005; 42(1):9-17. DOI: 10.1002/mc.20061 PubMed: 15486947
163. Smith-Oliver T, Butterworth BE. Correlation of the carcinogenic potential of di(2-ethylhexyl)phthalate (DEHP) with induced hyperplasia rather than with genotoxic activity. Mutat Res. 1987; 188(1):21-28. DOI: 10.1016/0165-1218(87)90110-8 PubMed: 3574334
164. Singh AR, Lawrence WH, Autian J. Mutagenic and antifertility sensitivities of mice to di-2-ethylhexyl phthalate (DEHP) and dimethoxyethyl phthalate (DMEP). Toxicol Appl Pharmacol. 1974; 29(1):35-46. DOI: 10.1016/0041-008X(74)90159-8 PubMed: 4283679
165. Jäckh R, Rhodes C, Grasso P, Carter JT. Genotoxicity studies on di-(2-ethylhexyl) phthalate and adipate and toxicity studies on di-(2-ethylhexyl) phthalate in the rat and marmoset. Food Chem Toxicol. 1984; 22(2):151-155. DOI: 10.1016/0278-6915(84)90096-6 PubMed: 6421684
166. Maronpot RR, Boorman GA. Interpretation of rodent hepatocellular proliferative alterations and hepatocellular tumors in chemical safety assessment. Toxicol Pathol. 1982; 10(2):71-78. DOI: 10.1177/019262338201000210 PubMed: 28094716
167. Boorman GA, Haseman JK, Waters MD, Hardisty JF, Sills RC. Quality review procedures necessary for rodent pathology databases and toxicogenomic studies: The National Toxicology Program experience. Toxicol Pathol. 2002; 30(1):88-92. DOI: 10.1080/01926230252824752 PubMed: 11890481
168. McConnell EE, Solleveld HA, Swenberg JA, Boorman GA. Guidelines for combining neoplasms for evaluation of rodent carcinogenesis studies. J Natl Cancer Inst. 1986; 76(2):283-289. PubMed: 3456066
169. U.S. Environmental Protection Agency (USEPA). Benchmark Dose Software (BMDS). Version 3.1.2. 2020. https://www.epa.gov/bmds
170. U.S. Environmental Protection Agency (USEPA). Benchmark dose software (BMDS) version 3.1.1. user guide. July 2019. 2019. EPA/600/R-18/310.
171. Kaplan EL, Meier P. Nonparametric estimation from incomplete observations. J Am Stat Assoc. 1958; 53(282):457-481. DOI: 10.1080/01621459.1958.10501452
172. Tarone RE. Tests for trend in life table analysis. Biometrika. 1975; 62(3):679-690. DOI: 10.1093/biomet/62.3.679
173. Cox DR. Regression models and life‐tables. J R Stat Soc Series B Stat Methodol. 1972; 34(2):187-202.
174. Bailer AJ, Portier CJ. Effects of treatment-induced mortality and tumor-induced mortality on tests for carcinogenicity in small samples. Biometrics. 1988; 44(2):417-431. DOI: 10.2307/2531856 PubMed: 3390507
175. Piegorsch W, Bailer A. Statistics for Environmental Biology and Toxicology: Section 6.3.2. London, UK: Chapman and Hall; 1997.
176. Portier CJ, Bailer AJ. Testing for increased carcinogenicity using a survival-adjusted quantal response test. Fundam Appl Toxicol. 1989; 12(4):731-737. DOI: 10.1016/0272-0590(89)90004-3 PubMed: 2744275
177. Portier CJ, Hedges JC, Hoel DG. Age-specific models of mortality and tumor onset for historical control animals in the National Toxicology Program’s carcinogenicity experiments. Cancer Res. 1986; 46(9):4372-4378. PubMed: 3731095
178. Bieler GS, Williams RL. Ratio estimates, the delta method, and quantal response tests for increased carcinogenicity. Biometrics. 1993; 49(3):793-801. DOI: 10.2307/2532200 PubMed: 8241374
179. Nam JM. A simple approximation for calculating sample sizes for detecting linear trend in proportions. Biometrics. 1987; 43(3):701-705. DOI: 10.2307/2532006 PubMed: 3663825
180. Rao JN, Scott AJ. A simple method for the analysis of clustered binary data. Biometrics. 1992; 48(2):577-585. DOI: 10.2307/2532311 PubMed: 1637980
181. Fung KY, Krewski D, Rao JN, Scott AJ. Tests for trend in developmental toxicity experiments with correlated binary data. Risk Anal. 1994; 14(4):639-648. DOI: 10.1111/j.1539-6924.1994.tb00277.x PubMed: 7972964
182. Gart JJ, Chu KC, Tarone RE. Statistical issues in interpretation of chronic bioassay tests for carcinogenicity. J Natl Cancer Inst. 1979; 62(4):957-974. PubMed: 285297
183. Dixon W, Massey F. Introduction to Statistical Analysis. New York, NY: McGraw Hill Book Company Inc; 1957. DOI: 10.2307/2332898
184. Tukey J. Easy summaries – numerical and graphical. Exploratory Data Analysis. Reading, MA: Addison-Wesley; 1977. p. 43-44.
185. Dunnett CW. A multiple comparison procedure for comparing several treatments with a control. J Am Stat Assoc. 1955; 50(272):1096-1121. DOI: 10.1080/01621459.1955.10501294
186. Williams DA. A test for differences between treatment means when several dose levels are compared with a zero dose control. Biometrics. 1971; 27(1):103-117. DOI: 10.2307/2528930 PubMed: 5547548
187. Williams DA. The comparison of several dose levels with a zero dose control. Biometrics. 1972; 28(2):519-531. DOI: 10.2307/2556164 PubMed: 5037867
188. Shirley E. A non-parametric equivalent of Williams’ test for contrasting increasing dose levels of a treatment. Biometrics. 1977; 33(2):386-389. DOI: 10.2307/2529789 PubMed: 884197
189. Williams DA. A note on Shirley’s nonparametric test for comparing several dose levels with a zero-dose control. Biometrics. 1986; 42(1):183-186. DOI: 10.2307/2531254 PubMed: 3719054
190. Dunn OJ. Multiple comparisons using rank sums. Technometrics. 1964; 6(3):241-252. DOI: 10.1080/00401706.1964.10490181
191. Jonckheere AR. A distribution-free k-sample test against ordered alternatives. Biometrika. 1954; 41(1-2):133-145. DOI: 10.1093/biomet/41.1-2.133
192. Hsu JC. The factor analytic approach to simultaneous inference in the general linear model. J Comput Graph Stat. 1992; 1(2):151-168. DOI: 10.1080/10618600.1992.10477011
193. Haseman JK. Data analysis: Statistical analysis and use of historical control data. Regul Toxicol Pharmacol. 1995; 21(1):52-59, discussion 81-56. DOI: 10.1006/rtph.1995.1009 PubMed: 7784636
194. Haseman JK, Rao GN. Effects of corn oil, time-related changes, and inter-laboratory variability on tumor occurrence in control Fischer 344 (F344/N) rats. Toxicol Pathol. 1992; 20(1):52-60. DOI: 10.1177/019262339202000107 PubMed: 1411131
195. Haseman JK. Value of historical controls in the interpretation of rodent tumor data. Drug Inf J. 1992; 26(2):191-200. DOI: 10.1177/009286159202600210
196. Code of Federal Regulations (CFR). 21(Part 58).
197. Miller JA, Miller EC. Ultimate chemical carcinogens as reactive mutagenic electrophiles. In: Hiatt HH, Watson JD, Winsten JA, editors. Origins of Human Cancer. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory; 1977. p. 605-627.
198. Straus DS. Somatic mutation, cellular differentiation, and cancer causation. J Natl Cancer Inst. 1981; 67:233-241. PubMed: 7021938
199. Crawford BD. Perspectives on the somatic mutation model of carcinogenesis. In: Mehlman MA, Flamm WG, Lorentzen RJ, editors. Advances in Modern Environmental Toxicology Mechanisms and Toxicity of Chemical Carcinogens and Mutagens. Princeton, NJ: Princeton Scientific Publishing Co. Inc; 1985. p. 13-59.
200. Ashby J, Tennant RW. Definitive relationships among chemical structure, carcinogenicity and mutagenicity for 301 chemicals tested by the US NTP. Mutat Res. 1991; 257(3):229-306. DOI: 10.1016/0165-1110(91)90003-E PubMed: 1707500
201. Witt KL, Knapton A, Wehr CM, Hook GJ, Mirsalis J, Shelby MD, MacGregor JT. Micronucleated erythrocyte frequency in peripheral blood of B6C3F(1) mice from short-term, prechronic, and chronic studies of the NTP carcinogenesis bioassay program. Environ Mol Mutagen. 2000; 36(3):163-194. DOI: 10.1002/1098-2280(2000)36:3<163::AID-EM1>3.0.CO;2-P PubMed: 11044899
202. National Toxicology Program (NTP). TR-601: Technical report pathology tables and curves pathology tables, survival and growth curves from NTP long term, genetic toxicology, and bench mark dose analyses of neoplastic lesions studies. Research Triangle Park, NC: U.S. Department of Health and Human Services, National Institute of Environmental Health Sciences, National Toxicology Program; 2020. DOI: 10.22427/NTP-DATA-TR-601
203. Araki A, Mitsui T, Miyashita C, Nakajima T, Naito H, Ito S, Sasaki S, Cho K, Ikeno T, Nonomura K, et al. Association between maternal exposure to di(2-ethylhexyl) phthalate and reproductive hormone levels in fetal blood: The Hokkaido study on environment and children’s health. PLoS One. 2014; 9(10):e109039. DOI: 10.1371/journal.pone.0109039 PubMed: 25296284
204. Latini G, De Felice C, Presta G, Del Vecchio A, Paris I, Ruggieri F, Mazzeo P. Exposure to di(2-ethylhexyl)phthalate in humans during pregnancy. Biol Neonate. 2003; 83(1):22-24. DOI: 10.1159/000067012 PubMed: 12566679
205. Li X, Sun H, Yao Y, Zhao Z, Qin X, Duan Y, Wang L. Distribution of phthalate metabolites between paired maternal–fetal samples. Environ Sci Technol. 2018; 52(11):6626-6635. DOI: 10.1021/acs.est.8b00838 PubMed: 29754483
206. Mose T, Knudsen LE, Hedegaard M, Mortensen GK. Transplacental transfer of monomethyl phthalate and mono(2-ethylhexyl) phthalate in a human placenta perfusion system. Int J Toxicol. 2007; 26(3):221-229. DOI: 10.1080/10915810701352721 PubMed: 17564903
207. Dalgaard M, Østergaard G, Lam HR, Hansen EV, Ladefoged O. Toxicity study of di(2-ethylhexyl)phthalate (DEHP) in combination with acetone in rats. Pharmacol Toxicol. 2000; 86(2):92-100. DOI: 10.1034/j.1600-0773.2000.pto860208.x PubMed: 10728921
208. Dalsenter PR, Santana GM, Grande SW, Andrade AJ, Araujo SL. Phthalate affect the reproductive function and sexual behavior of male Wistar rats. Hum Exp Toxicol. 2006; 25(6):297-303. DOI: 10.1191/0960327105ht624oa PubMed: 16866186
209. Pocar P, Fiandanese N, Secchi C, Berrini A, Fischer B, Schmidt JS, Schaedlich K, Borromeo V. Exposure to di(2-ethyl-hexyl) phthalate (DEHP) in utero and during lactation causes long-term pituitary-gonadal axis disruption in male and female mouse offspring. Endocrinology. 2012; 153(2):937-948. DOI: 10.1210/en.2011-1450 PubMed: 22147016
210. National Toxicology Program (NTP). Diethylhexylphthalate: Multigenerational reproductive assessment by continuous breeding when administered to Sprague-Dawley rats in the diet. Research Triangle Park, NC: U.S. Department of Health and Human Services, National Institute of Environmental Health Sciences, National Toxicology Program; 2005. PB2005107575.
211. National Toxicology Program (NTP). NTP technical report on the toxicology and carcinogenesis studies of perfluorooctanoic acid (CASRN 335-67-1) administered in feed to Sprague Dawley (Hsd:Sprague Dawley SD) rats. Research Triangle Park, NC: U.S. Department of Health and Human Services, National Institute of Environmental Health Sciences, National Toxicology Program; 2020. NTP Technical Report No. 598. https://ntp.niehs.nih.gov/publications/reports/tr/500s/tr598/index.html?utm_source=direct&utm_medium=prod&utm_campaign=ntpgolinks&utm_term=tr598abs PubMed: 33556048
212. Rusyn I, Corton JC. Mechanistic considerations for human relevance of cancer hazard of di(2-ethylhexyl) phthalate. Mutat Res. 2012; 750(2):141-158. DOI: 10.1016/j.mrrev.2011.12.004 PubMed: 22198209
213. National Toxicology Program (NTP). NTP toxicology and carcinogenesis studies of butyl benzyl phthalate (CAS No. 85-68-7) in F334/N rats (Feed studies). Research Triangle Park, NC: U.S. Department of Health and Human Services, National Institute of Environmental Health Sciences, National Toxicology Program; 1997. NTP Technical Report No. 458. https://ntp.niehs.nih.gov/publications/reports/tr/400s/tr458/index.html?utm_source=direct&utm_medium=prod&utm_campaign=ntpgolinks&utm_term=tr458abs PubMed: 12587018
214. Biegel LB, Hurtt ME, Frame SR, O’Connor JC, Cook JC. Mechanisms of extrahepatic tumor induction by peroxisome proliferators in male CD rats. Toxicol Sci. 2001; 60(1):44-55. DOI: 10.1093/toxsci/60.1.44 PubMed: 11222872
215. Bell RH Jr, Kuhlmann ET, Jensen RT, Longnecker DS. Overexpression of cholecystokinin receptors in azaserine-induced neoplasms of the rat pancreas. Cancer Res. 1992; 52(12):3295-3299. PubMed: 1596887
216. Longnecker DS. Interface between adaptive and neoplastic growth in the pancreas. Gut. 1987; 28 Suppl:253-258. DOI: 10.1136/gut.28.Suppl.253 PubMed: 3319812
217. Povoski SP, Zhou W, Longnecker DS, Roebuck BD, Bell RH Jr. Stimulation of growth of azaserine-induced putative preneoplastic lesions in rat pancreas is mediated specifically by way of cholecystokinin-A receptors. Cancer Res. 1993; 53(17):3925-3929. PubMed: 8358719
218. Gray LE, Foster PMD. Significance of experimental studies for assessing adverse effects of endocrine-disrupting chemicals. Pure Appl Chem. 2003; 75(11-12):2125-2141. DOI: 10.1351/pac200375112125
219. Gray LE Jr, Furr J, Tatum-Gibbs KR, Lambright C, Sampson H, Hannas BR, Wilson VS, Hotchkiss A, Foster PM. Establishing the “biological relevance” of dipentyl phthalate reductions in fetal rat testosterone production and plasma and testis testosterone levels. Toxicol Sci. 2016; 149(1):178-191. DOI: 10.1093/toxsci/kfv224 PubMed: 26454885
220. Howdeshell KL, Furr J, Lambright CR, Rider CV, Wilson VS, Gray LE Jr. Cumulative effects of dibutyl phthalate and diethylhexyl phthalate on male rat reproductive tract development: Altered fetal steroid hormones and genes. Toxicol Sci. 2007; 99(1):190-202. DOI: 10.1093/toxsci/kfm069 PubMed: 17400582
221. Borch J, Axelstad M, Vinggaard AM, Dalgaard M. Diisobutyl phthalate has comparable anti-androgenic effects to di-n-butyl phthalate in fetal rat testis. Toxicol Lett. 2006; 163(3):183-190. DOI: 10.1016/j.toxlet.2005.10.020 PubMed: 16458459
222. Welsh M, Saunders PTK, Fisken M, Scott HM, Hutchison GR, Smith LB, Sharpe RM. Identification in rats of a programming window for reproductive tract masculinization, disruption of which leads to hypospadias and cryptorchidism. J Clin Invest. 2008; 118(4):1479-1490. DOI: 10.1172/JCI34241 PubMed: 18340380
223. Nef S, Parada LF. Cryptorchidism in mice mutant for Insl3. Nat Genet. 1999; 22(3):295-299. DOI: 10.1038/10364 PubMed: 10391220
224. Zimmermann S, Steding G, Emmen JMA, Brinkmann AO, Nayernia K, Holstein AF, Engel W, Adham IM. Targeted disruption of the Insl3 gene causes bilateral cryptorchidism. Mol Endocrinol. 1999; 13(5):681-691. DOI: 10.1210/mend.13.5.0272 PubMed: 10319319
225. Wilson VS, Lambright C, Furr J, Ostby J, Wood C, Held G, Gray LE. Phthalate ester-induced gubernacular lesions are associated with reduced insl3 gene expression in the fetal rat testis. Toxicol Lett. 2004; 146(3):207-215. DOI: 10.1016/j.toxlet.2003.09.012 PubMed: 14687758
226. Barlow NJ, McIntyre BS, Foster PMD. Male reproductive tract lesions at 6, 12, and 18 months of age following in utero exposure to di(n-butyl) phthalate. Toxicol Pathol. 2004; 32(1):79-90. DOI: 10.1080/01926230490265894 PubMed: 14713552
227. Furr JR, Lambright CS, Wilson VS, Foster PM, Gray LE Jr. A short-term in vivo screen using fetal testosterone production, a key event in the phthalate adverse outcome pathway, to predict disruption of sexual differentiation. Toxicol Sci. 2014; 140(2):403-424. DOI: 10.1093/toxsci/kfu081 PubMed: 24798384
228. Fisher JS, Macpherson S, Marchetti N, Sharpe RM. Human ‘testicular dysgenesis syndrome’: A possible model using in-utero exposure of the rat to dibutyl phthalate. Hum Reprod. 2003; 18(7):1383-1394. DOI: 10.1093/humrep/deg273 PubMed: 12832361
229. van den Driesche S, Kilcoyne KR, Wagner I, Rebourcet D, Boyle A, Mitchell R, McKinnell C, Macpherson S, Donat R, Shukla CJ, et al. Experimentally induced testicular dysgenesis syndrome originates in the masculinization programming window. JCI Insight. 2017; 2(6):e91204. DOI: 10.1172/jci.insight.91204 PubMed: 28352662
230. Sohval AR. Testicular dysgenesis as an etiologic factor in cryptorchidism. J Urol. 1954; 72(4):693-702. DOI: 10.1016/S0022-5347(17)67649-3 PubMed: 13202269
231. Sohval AR. Testicular dysgenesis in relation to neoplasm of the testicle. J Urol. 1956; 75(2):285-291. DOI: 10.1016/S0022-5347(17)66809-5 PubMed: 13296128
232. Hoei-Hansen CE, Holm M, Rajpert-De Meyts E, Skakkebaek NE. Histological evidence of testicular dysgenesis in contralateral biopsies from 218 patients with testicular germ cell cancer. J Pathol. 2003; 200(3):370-374. DOI: 10.1002/path.1372 PubMed: 12845633
233. Gray TJB, Butterworth KR, Gaunt IF, Grasso P, Gangolli SD. Short-term toxicity study of di-(2-ethylhexyl) phthalate in rats. Food Cosmet Toxicol. 1977; 15(5):389-399. DOI: 10.1016/S0015-6264(77)80003-5 PubMed: 598790
234. Clegg ED, Cook JC, Chapin RE, Foster PM, Daston GP. Leydig cell hyperplasia and adenoma formation: Mechanisms and relevance to humans. Reprod Toxicol. 1997; 11(1):107-121. DOI: 10.1016/S0890-6238(96)00203-1 PubMed: 9138629
235. Cook JC, Klinefelter GR, Hardisty JF, Sharpe RM, Foster PM. Rodent Leydig cell tumorigenesis: A review of the physiology, pathology, mechanisms, and relevance to humans. Crit Rev Toxicol. 1999; 29(2):169-261. DOI: 10.1080/10408449991349203 PubMed: 10213111
236. Creasy D, Bube A, de Rijk E, Kandori H, Kuwahara M, Masson R, Nolte T, Reams R, Regan K, Rehm S, et al. Proliferative and nonproliferative lesions of the rat and mouse male reproductive system. Toxicol Pathol. 2012; 40(6 Suppl):40S-121S. DOI: 10.1177/0192623312454337 PubMed: 22949412
237. Wei Z, Song L, Wei J, Chen T, Chen J, Lin Y, Xia W, Xu B, Li X, Chen X, et al. Maternal exposure to di-(2-ethylhexyl)phthalate alters kidney development through the renin-angiotensin system in offspring. Toxicol Lett. 2012; 212(2):212-221. DOI: 10.1016/j.toxlet.2012.05.023 PubMed: 22677342
238. Khorana AA, Fine RL. Pancreatic cancer and thromboembolic disease. Lancet Oncol. 2004; 5(11):655-663. DOI: 10.1016/S1470-2045(04)01606-7 PubMed: 15522652
239. Kamijo Y, Hora K, Nakajima T, Kono K, Takahashi K, Ito Y, Higuchi M, Kiyosawa K, Shigematsu H, Gonzalez FJ, et al. Peroxisome proliferator-activated receptor alpha protects against glomerulonephritis induced by long-term exposure to the plasticizer di-(2-ethylhexyl)phthalate. J Am Soc Nephrol. 2007; 18(1):176-188. DOI: 10.1681/ASN.2006060597 PubMed: 17135395
240. Organisation for Economic Co-operation and Development (OECD). OECD guidelines for testing of chemicals. Test guideline 479. Test no. 479: Genetic toxicology: In vitro sister chromatid exchange assay in mammalian cells. 2014. https://www.oecd-ilibrary.org/environment/test-no-479-genetic-toxicology-in-vitro-sister-chromatid-exchange-assay-in-mammalian-cells_9789264071384-en
241. McFee AF, Lowe KW, San Sebastian JR. Improved sister-chromatid differentiation using paraffin-coated bromodeoxyuridine tablets in mice. Mutat Res. 1983; 119(1):83-88. DOI: 10.1016/0165-7992(83)90042-8 PubMed: 6823230
242. McFee AF, Tice RR, Shelby MD. In vivo cytogenetic activity of diphenylhydantoin in mice. Mutat Res. 1992; 278(1):61-68. DOI: 10.1016/0165-1218(92)90286-9 PubMed: 1370120
243. Shelby MD, Gulati DK, Tice RR, Wojciechowski JP. Results of tests for micronuclei and chromosomal aberrations in mouse bone marrow cells with the human carcinogens 4-aminobiphenyl, treosulphan, and melphalan. Environ Mol Mutagen. 1989; 13(4):339-342. DOI: 10.1002/em.2850130410 PubMed: 2737185
244. Margolin BH, Resnick MA, Rimpo JY, Archer P, Galloway SM, Bloom AD, Zeiger E. Statistical analyses for in vitro cytogenetic assays using Chinese hamster ovary cells. Environ Mutagen. 1986; 8(2):183-204. DOI: 10.1002/em.2860080203 PubMed: 3698942
245. MacGregor JT, Wehr CM, Henika PR, Shelby MD. The in vivo erythrocyte micronucleus test: Measurement at steady state increases assay efficiency and permits integration with toxicity studies. Fundam Appl Toxicol. 1990; 14(3):513-522. DOI: 10.1016/0272-0590(90)90255-I PubMed: 2111256
Conclusions
Under the conditions of the perinatal and postweaning feed study (Study 1), there was clear evidence of carcinogenic activity of di(2-ethylhexyl) phthalate (DEHP) in male Hsd:Sprague Dawley SD rats based on the increased incidences of hepatocellular adenoma or carcinoma (combined) and acinar adenoma or carcinoma (combined) neoplasms (predominately adenomas) of the pancreas. There was clear evidence of carcinogenic activity of DEHP in female Hsd:Sprague Dawley SD rats based on the increased incidence of hepatocellular adenoma or carcinoma (combined). The occurrence of pancreatic acinar adenoma or carcinoma (combined) was considered to be related to exposure. The occurrence of uterine (including cervix) adenoma, adenocarcinoma, squamous cell carcinoma, or squamous cell papilloma (combined) in female rats may have been related to exposure.
Under the conditions of the postweaning-only feed study (Study 2), there was clear evidence of carcinogenic activity of DEHP in male Hsd:Sprague Dawley SD rats based on the increased incidences of hepatocellular adenoma or carcinoma (combined) and acinar adenoma or carcinoma (combined) neoplasms (predominately adenomas) of the pancreas. The occurrence of testicular interstitial cell adenoma in male rats may have been related to exposure. There was clear evidence of carcinogenic activity of DEHP in female Hsd:Sprague Dawley SD rats based on the increased incidences of hepatocellular adenoma or carcinoma (combined) and uterine (including cervix) adenoma, adenocarcinoma, squamous cell carcinoma, or squamous cell papilloma (combined). The occurrence of pancreatic acinar adenoma or carcinoma (combined) in female rats was considered to be related to exposure.
The BMD analysis shows there was no consistent pattern indicating that perinatal and postweaning exposure was more sensitive compared to postweaning-only exposure and modeled responses were within threefold of each other. However, there was a stronger carcinogenic response in the reproductive organs (uterus and testis) in the postweaning-only exposure study compared to the perinatal and postweaning exposure study.
Perinatal and postweaning exposure to DEHP (Study 1) resulted in increased incidence of nonneoplastic lesions in the liver, kidney, heart (male), pancreas (female), pituitary gland (male), bone marrow (male), testis, and epididymis. In addition, exposure increased gross lesions within the reproductive tract of males and females.
Postweaning exposure to DEHP (Study 2) resulted in increased incidence of nonneoplastic lesions in the liver, pancreas, heart (male), pituitary gland (male), bone marrow (male), testis, epididymis, and uterus.
Appendices
Appendix A. Chemical Characterization and Dose Formulation Studies
A.1.
Procurement and Characterization of Di(2-ethylhexyl) Phthalate
Di(2-ethylhexyl) phthalate (DEHP) was obtained from Aldrich Chemical Company Inc. (St. Louis, MO) in a single lot (lot 01514TH) received in two shipments. The first shipment (10 L) was received on December 12, 2008 and used for chemical characterization. The second shipment (250 L) was received on November 4, 2009 and used for the dose formulations in the 2-year studies and chemical reanalysis. Identity, purity, and stability analyses were conducted by the analytical chemistry laboratory at RTI International (Research Triangle Park, NC). Reports on analyses performed in support of the DEHP studies are on file at the National Institute of Environmental Health Sciences.
The appearance (clear liquid) and density of lot 01514TH (0.976 g/mL at 21.9°C) matched that of DEHP (0.985 g/mL at 25°C). Galbraith Laboratories (Knoxville, TN) performed the boiling point and elemental analyses of lot 01514TH. While the elemental analysis confirmed the anticipated relative ratios, the experimental boiling point (330.9°C) was considerably lower than that reported in the literature (384°C). Using a different method, the results (335°C, 760 mm Hg) from RTI International were consistent with Galbraith Laboratories. A precise molecular mass was measured using research-grade high-resolution mass spectrometry (HRMS) at the University of South Carolina Mass Spectrometry Facility (Columbia, SC). The observed mass values (390.2772) were within acceptable limits (≤5 ppm) of the calculated mass (390.2770).
The identity of lot 01514TH was confirmed using infrared (IR) spectroscopy, 1H and 13C nuclear magnetic resonance (NMR) spectroscopy, and gas chromatography (GC) with MS detection. The IR spectrum was in good agreement with the structure of DEHP and with the reference spectrum from the National Institute of Advanced Industrial Science and Technology Spectral Database for Organic Compounds (SDBS No. 2266) for DEHP (Figure A-1). 1H and 13C NMR spectra (Figure A-2, Figure A-3) were consistent with the structure of DEHP and the prediction from the Advanced Chemistry Development Spectral Prediction Program (Version 10.02, Toronto, Ontario, Canada) for DEHP. GC/MS identified the major peak from the 10 L shipment of lot 01514TH as DEHP using fragmentation pattern analysis and comparison with the National Institute of Standards and Technology (NIST) reference spectrum (No. 311338) for DEHP (Table A-1, System A). The GC/MS spectra correlated well with the structure of DEHP.
The moisture content of lot 01514TH was determined by Karl Fisher titration. The purity was determined using ultra-performance liquid chromatography (UPLC) with photodiode array detector (PDA) and using GC with flame ionization detection (FID) (Table A-1, Systems B and C, respectively). The Karl Fisher titration yielded a water content of 0.145%. UPLC/PDA analysis demonstrated one major peak accounting for 99.7% and one minor peak accounting for 0.2% of the total integrated area. GC/FID analysis also found one major peak accounting for 99.7% and one minor peak accounting for 0.3% of the total integrated area. An additional GC/MS analysis of the test chemical was performed in an attempt to identify the minor component in the chromatographic profile (Table A-1, System A). The fragmentation of the minor impurity peak agreed with the NIST reference spectrum (No. 312137) for mono(2-ethylhexyl) phthalate.
Accelerated stability studies were conducted by the analytical chemistry laboratory using samples of lot 01514TH stored at ambient temperature (approximately 22°C), refrigerated temperature (approximately 5°C), and elevated temperature (approximately 60°C) in amber vials sealed with foil-lined caps. After 14 days, samples were analyzed by GC/FID (Table A-1, System C). Stability of DEHP was confirmed for at least 2 weeks when stored in sealed glass vials at temperatures from 5°C to 60°C.
Upon receipt of the 250 L shipment used for the 2-year studies, the bulk chemical of lot 01514TH was homogenized by shaking each of the 5 50 L plastic jugs for approximately 2 minutes and transferred to 70 4 L amber glass storage bottles, which were stored at room temperature.
Prior to using the bulk chemical from the 250 L shipment of lot 01514TH for dose formulations, the identity was confirmed using the same GC/MS system with comparison to the reference spectrum and an aliquot of the test article from the 10L shipment (Table A-1, System A). The GC/MS analysis of the 250 L shipment of lot 01514TH demonstrated one major peak accounting for 99.9% of the total integrated area. Periodic reanalysis of the bulk chemical lot 01514TH was performed prior to and during the animal studies by the laboratory using high-performance liquid chromatography (HPLC) with ultraviolet (UV) detection (Table A-1, System D), and no degradation of the test chemical was detected.
A.2.
Preparation and Analysis of Dose Formulations
The base diet was meal feed purchased from Zeigler Brothers, Inc. (Gardners, PA). The perinatal and postweaning study (Study 1) utilized NIH-07 feed (2 lots milled March and April 2011) in addition to NTP-2000 feed (25 lots milled April 2011–March 2013). The postweaning-only study (Study 2) utilized NTP-2000 feed (25 lots milled December 2010–December 2012). In addition to determining the suitability of the vehicles for feeding the animals, analysis of the NTP-2000 feed extract performed by the study laboratory using liquid chromatography mass spectrometry (LC/MS) confirmed that the vehicle did not contain the test article DEHP.
Dose formulations were prepared monthly by mixing DEHP with feed (Table A-2). For the perinatal and postweaning study, formulations were prepared at concentrations of 0, 300, 1,000, 3,000, and 10,000 ppm in both NIH-07 feed (May 4, May 24, and June 15, 2011) and in NTP-2000 feed (31 formulations; June 2011–June 2013). For the postweaning-only study, formulations were prepared in NTP-2000 feed at concentrations of 0, 300, 1,000, 3,000, and 10,000 ppm (31 formulations; February 2011–February 2013). The plastic bags used by the study laboratory in the preparation and storage of blank and dosed feed were determined to have no levels of DEHP above the limit of detection of the assay (1.27 ppm).
Homogeneity studies were performed on the 25 and 10,000 ppm dose formulations in both 25-kg NIH-07 feed batch sizes and 25-kg NTP-2000 feed batch sizes by the analytical chemistry laboratory using UPLC/PDA (Table A-1, System B). Additional homogeneity studies of the 300 and 10,000 ppm dose formulations in a 72-kg NIH-07 feed batch size and 300, 3,000, and 10,000 ppm dose formulations in a 92-kg NTP-2000 feed batch sizes were performed before the animal studies by the study laboratory using HPLC/UV (Table A-1, System D). All formulations analyzed were determined to be homogenous and of appropriate concentration.
Stability studies conducted by the chemistry laboratory of the 25 ppm NIH-07 and 25 ppm NTP-2000 dose formulations confirmed the stability of DEHP after 42 days at room, refrigerated, or frozen temperatures. Stability was also confirmed under simulated dosing conditions (room temperature, exposure to air and light for 7 days, in absence of excreta, and in presence of excreta). Control and dosed formulations were stored in individual plastic bag-lined containers at room temperature (approximately 25°C) and were used within 42 days of preparation.
Periodic analyses of the preadministration dose formulations of DEHP were conducted by the study laboratory every 1 to 3 months to determine purity, while postadministration (animal room) samples were analyzed about every 1 to 7 months (Table A-3, Table A-4). All preadministration formulations were within 10% of the target concentrations. For the perinatal and postweaning study, all postadministration dose formulations of DEHP were within 10% of target concentrations. For the postweaning-only study (Study 2), all postadministration dose formulations of DEHP were within 10% of target concentrations except for the 1,000 ppm dose formulation prepared on July 30, 2012, collected from residual feed in the feeder that was 12.3% below the target concentration.
Table A-1. Chromatography Systems Used in the Two-year Feed Studies of Di(2-ethyhexyl) Phthalate
| Chromatography | Detection System | Column | Mobile Phase |
|---|---|---|---|
| System A | |||
| Gas chromatography | Mass selective detector | J&W DB-1 (25 m × 0.32 mm ID, 0.25 μm film thickness) | Helium, 1.65 mL/min flow rate |
| System B | |||
| Ultra-performance liquid chromatography | Photodiode array detector (205 to 400 nm, extracted at 225 nm) | Waters Acquity UPLC BEH Phenyl (50 mm × 2.1 mm ID, 1.7 μm particle size), with Waters Acquity In-Line Filter (0.2 μm) | A: Methanol B: Water Gradient program: A:B 25:75 to 75:25 in 3 min, hold at 78:22 for 1 min, ramp to 100:0 in 1 min, hold at 100:0 for 1 min, reverse to 25:75 in 0.5 min, hold at 25:75 for 1.5 min 0.6 mL/min flow rate |
| System C | |||
| Gas chromatography | Flame ionization detection (325°C) | J&W HP-5 (30 m × 0.32 mm ID, 0.25 μm film thickness) | Helium, 1 mL/min flow rate |
| System D | |||
| High-performance liquid chromatography | Ultraviolet (225 nm) | Thermo Scientific Hypersil Phenyl (250 mm × 4.6 mm ID, 5 μm particle size) with Hypersil Phenyl guard (5 μm particle size) | A: Methanol B: ASTM Type I Water Gradient program: A:B 70:30 to 85:15 in 5 min, ramp to 100:0 in 4 min, hold at 100:0 for 4 min, reverse to 70:30 in 0.1 min, hold at 70:30 for 10.9 min 1.0 mL/min flow rate |
Table A-2. Preparation and Storage of Dose Formulations in the Two-year Feed Studies of Di(2-ethylhexyl) Phthalate
| Preparation |
|---|
| Stock solutions of di(2-ethylhexyl) phthalate (DEHP) were created by weighing an appropriate amount of lot 01514TH and adding it to a volumetric flask. Acetone was used to bring the solution to volume. Flasks of stocks solution were sealed and shaken until the chemical was dissolved (at least 10 inversions). An initial formulation premix was created by weighing an appropriate amount of feed (NIH-07 or NTP-2000) into a mixing bowl. A portion of the stock DEHP solution was slowly poured onto the feed and then stirred for 2 minutes at a low setting using a Hobart mixer. The mixer was stopped, and the remaining stock solution was poured onto the feed. The stock container was rinsed with acetone twice and the rinses were poured onto the feed. The premix feed was stirred under a nitrogen stream with a flow rate of 10 liters per minute for approximately an hour to encourage cyclonic flow and to ensure complete evaporation of the acetone. The formulation blends were prepared by adding half of the required blank feed to a twin shell blender and then evenly covering with the premix. The sides were “rinsed” twice with the remaining blank feed and added to the blender. The final formulation was mixed in the blender for 15 minutes. The dose formulations were prepared approximately every 4 weeks. |
| Chemical lot number |
| 01514TH |
| Maximum storage time |
| 42 days |
| Storage conditions |
| Stored in sealed plastic bag-lined container at ~25°C |
| Study laboratory |
| Battelle (Columbus, OH) |
Table A-3. Results of Analyses of Dose Formulations Administered to Rats in the Perinatal and Postweaning Two-year Feed Study of Di(2-ethylhexyl) Phthalate
| Date Prepared | Date Analyzed | Target Concentration (ppm) |
Determined Concentration (ppm)a | Difference from Target (%) |
|---|---|---|---|---|
| May 4, 2011 | May 5, 2011 | 0 | BLOQ | NA |
| 300 | 309 ± 5 | 3.0 | ||
| 1,000 | 1,020 ± 20 | 2.0 | ||
| 3,000 | 3,170 ± 70 | 5.7 | ||
| 10,000 | 10,200 ± 100 | 2.0 | ||
| June 6, 2011 | June 6, 2011 | 0 | BLOQ | NA |
| 300 | 291.5 | −2.8 | ||
| 1,000 | 959.5 | −4.1 | ||
| 3,000 | 2,885 | −3.8 | ||
| 10,000 | 9,790 | −2.1 | ||
| August 17, 2011 | August 19, 2011 | 0 | BLOQ | NA |
| 300 | 292.5 | −2.5 | ||
| 1,000 | 982.5 | −1.8 | ||
| 3,000 | 2,930 | −2.3 | ||
| 10,000 | 9,805 | −2.0 | ||
| October 31, 2011 | November 3, 2011 | 0 | BLOQ | NA |
| 300 | 287.5 | −4.2 | ||
| 1,000 | 922 | −7.8 | ||
| 3,000 | 2,875 | −4.2 | ||
| 10,000 | 9,560 | −4.4 | ||
| January 12, 2012 | January 13, 2012 | 0 | BLOQ | NA |
| 300 | 305 | 1.7 | ||
| 1,000 | 993 | −0.7 | ||
| 3,000 | 2,990 | −0.3 | ||
| 10,000 | 10,100 | 1.0 | ||
| March 26, 2012 | March 29, 2012 | 0 | BLOQ | NA |
| 300 | 307.5 | 2.5 | ||
| 1,000 | 1,020 | 2.0 | ||
| 3,000 | 3,045 | 1.5 | ||
| 10,000 | 10,350 | 3.5 | ||
| June 6, 2012 | June 7, 2012 | 0 | BLOQ | NA |
| 300 | 295 | −1.7 | ||
| 1,000 | 983 | −1.7 | ||
| 3,000 | 2,990 | −0.3 | ||
| 10,000 | 9,850 | −1.5 | ||
| July 30, 2012 | August 1, 2012 | 0 | BLOQ | NA |
| 300 | 300.5 | 0.2 | ||
| 1,000 | 997.5 | −0.3 | ||
| 3,000 | 3,040 | 1.3 | ||
| 10,000 | 10,200 | 2.0 | ||
| October 8, 2012 | October 8, 2012 | 0 | BLOQ | NA |
| 300 | 314.5 | 4.8 | ||
| 1,000 | 998 | −0.2 | ||
| 3,000 | 2,860 | −4.7 | ||
| 10,000 | 9,895 | −1.1 | ||
| December 18, 2012 | December 18, 2012 | 0 | BLOQ | NA |
| 300 | 292 | −2.7 | ||
| 1,000 | 971 | −2.9 | ||
| 3,000 | 3,015 | 0.5 | ||
| 10,000 | 9,845 | −1.6 | ||
| March 4, 2013 | March 5, 2013 | 0 | BLOQ | NA |
| 300 | 302 ± 2 | 0.7 | ||
| 1,000 | 998 ± 3 | −0.2 | ||
| 3,000 | 3,000 ± 40 | 0.0 | ||
| 10,000 | 10,000 ± 200 | 0.0 | ||
| April 22, 2013 | April 22, 2013 | 0 | BLOQ | NA |
| 300 | 299 ± 3 | −0.3 | ||
| 1,000 | 995 ± 2 | −0.5 | ||
| 3,000 | 2,970 ± 10 | −1.0 | ||
| 10,000 | 9,840 ± 30 | −1.6 | ||
| Animal room samples | ||||
| May 4, 2011 | June 13, 2011 (feeder) | 0 | BLOQ | NA |
| 300 | 279 ± 1 | −6.9 | ||
| 1,000 | 939 ± 3 | −6.1 | ||
| 3,000 | 2,870 ± 30 | −4.5 | ||
| 10,000 | 9,480 ± 50 | −5.2 | ||
| June 13, 2011 (bucket) | 0 | BLOQ | NA | |
| 300 | 294 ± 1 | −2.1 | ||
| 1,000 | 957 ± 24 | −4.3 | ||
| 3,000 | 2,920 ± 130 | −2.7 | ||
| 10,000 | 9,560 ± 120 | −4.4 | ||
| June 6, 2011 | July 19, 2011 (feeder) | 0 | BLOQ | NA |
| 300 | 295 ± 2 | −1.8 | ||
| 1,000 | 960 ± 1 | −4.0 | ||
| 3,000 | 2,880 ± 20 | −4.0 | ||
| 10,000 | 10,000 ± 0 | 0.0 | ||
| July 19, 2011 (bucket) | 0 | BLOQ | NA | |
| 300 | 302 ± 1 | 0.5 | ||
| 1,000 | 1,000 ± 10 | 0.3 | ||
| 3,000 | 3,000 ± 30 | 0.1 | ||
| 10,000 | 9,770 ± 120 | −0.3 | ||
| January 12, 2012 | February 23, 2012 (feeder) | 0 | BLOQ | NA |
| 300 | 298 ± 1 | −0.6 | ||
| 1,000 | 958 ± 5 | −4.2 | ||
| 3,000 | 2,750 ± 30 | −8.3 | ||
| 10,000 | 9,270 ± 120 | −7.3 | ||
| February 23, 2012 (bucket) | 0 | BLOQ | NA | |
| 300 | 297 ± 3 | −0.9 | ||
| 1,000 | 980 ± 6 | −2.0 | ||
| 3,000 | 2,900 ± 30 | −3.4 | ||
| 10,000 | 9,360 ± 130 | −6.4 | ||
| July 30, 2012 | September 5, 2012 (feeder) | 0 | BLOQ | NA |
| 300 | 288 ± 1 | −4.1 | ||
| 1,000 | 1,020 ± 60 | 2.2 | ||
| 3,000 | 2,840 ± 60 | −5.3 | ||
| 10,000 | 9,190 ± 110 | −8.1 | ||
| September 5, 2012 (bucket) | 0 | BLOQ | NA | |
| 300 | 288 ± 2 | −4.1 | ||
| 1,000 | 1,000 ± 80 | 0.0 | ||
| 3,000 | 2,830 ± 130 | −5.7 | ||
| 10,000 | 9,920 ± 280 | −0.8 | ||
| March 4, 2013 | April 15, 2013 (feeder) | 0 | BLOQ | NA |
| 300 | 281 ± 2 | −6.2 | ||
| 1,000 | 909 ± 4 | −9.1 | ||
| 3,000 | 2,740 ± 20 | −8.6 | ||
| 10,000 | 9,510 ± 20 | −4.9 | ||
| April 15, 2013 (bucket) | 0 | BLOQ | NA | |
| 300 | 289 ± 0 | −3.7 | ||
| 1,000 | NS | NA | ||
| 3,000 | 2,910 ± 70 | −3.0 | ||
| 10,000 | 10,200 ± 200 | 2.0 | ||
Table A-4. Results of Analyses of Dose Formulations Administered to Rats in the Postweaning-only Two-year Feed Study of Di(2-ethylhexyl) Phthalate
| Date Prepared | Date Analyzed | Target Concentration (ppm) |
Determined Concentration (ppm)a | Difference from Target (%) |
|---|---|---|---|---|
| February 2, 2011 | February 5, 2011 | 0 | BLOQ | NA |
| 300 | 295 ± 5 | −1.7 | ||
| 1,000 | 987 ± 38 | −1.3 | ||
| 3,000 | 3,040 ± 10 | 1.3 | ||
| 10,000 | 10,300 ± 300 | 3.0 | ||
| March 28, 2011 | March 28, 2011 | 0 | BLOQ | NA |
| 300 | 300 ± 3 | 0.0 | ||
| 1,000 | 991 ± 3 | −0.9 | ||
| 3,000 | 2,990 ± 10 | −0.3 | ||
| 10,000 | 9,880 ± 190 | −1.2 | ||
| June 6, 2011 | June 6, 2011 | 0 | BLOQ | NA |
| 300 | 291.5 | −2.8 | ||
| 1,000 | 959.5 | −4.1 | ||
| 3,000 | 2,885 | −3.8 | ||
| 10,000 | 9,790 | −2.1 | ||
| August 17, 2011 | August 19, 2011 | 0 | BLOQ | NA |
| 300 | 292.5 | −2.5 | ||
| 1,000 | 982.5 | −1.8 | ||
| 3,000 | 2,930 | −2.3 | ||
| 10,000 | 9,805 | −2.0 | ||
| October 31, 2011 | November 3, 2011 | 0 | BLOQ | NA |
| 300 | 287.5 | −4.2 | ||
| 1,000 | 922 | −7.8 | ||
| 3,000 | 2,875 | −4.2 | ||
| 10,000 | 9,560 | −4.4 | ||
| January 12, 2012 | January 13, 2012 | 0 | BLOQ | NA |
| 300 | 305 | 1.7 | ||
| 1,000 | 993 | −0.7 | ||
| 3,000 | 2,990 | −0.3 | ||
| 10,000 | 10,100 | 1.0 | ||
| March 26, 2012 | March 29, 2012 | 0 | BLOQ | NA |
| 300 | 307.5 | 2.5 | ||
| 1,000 | 1,020 | 2.0 | ||
| 3,000 | 3,045 | 1.5 | ||
| 10,000 | 10,350 | 3.5 | ||
| June 6, 2012 | June 7, 2012 | 0 | BLOQ | NA |
| 300 | 295 | −1.7 | ||
| 1,000 | 983 | −1.7 | ||
| 3,000 | 2,990 | −0.3 | ||
| 10,000 | 9,850 | −1.5 | ||
| July 30, 2012 | August 1, 2012 | 0 | BLOQ | NA |
| 300 | 300.5 | 0.2 | ||
| 1,000 | 997.5 | −0.3 | ||
| 3,000 | 3,040 | 1.3 | ||
| 10,000 | 10,200 | 2.0 | ||
| October 8, 2012 | October 8, 2012 | 0 | BLOQ | NA |
| 300 | 314.5 | 4.8 | ||
| 1,000 | 998 | −0.2 | ||
| 3,000 | 2,860 | −4.7 | ||
| 10,000 | 9,895 | −1.1 | ||
| December 18, 2012 | December 18, 2012 | 0 | BLOQ | NA |
| 300 | 292 | −2.7 | ||
| 1,000 | 971 | −2.9 | ||
| 3,000 | 3,015 | 0.5 | ||
| 10,000 | 9,845 | −1.6 | ||
| Animal room samples | ||||
| February 2, 2011 | March 17, 2011 (feeder) | 0 | BLOQ | NA |
| 300 | 293 ± 6 | −2.2 | ||
| 1,000 | 933 ± 4 | −6.7 | ||
| 3,000 | 2,830 ± 30 | −5.6 | ||
| 10,000 | 9,430 ± 280 | −5.7 | ||
| March 17, 2011 (bucket) | 0 | BLOQ | NA | |
| 300 | 294 ± 6 | −1.9 | ||
| 1,000 | 972 ± 11 | −2.8 | ||
| 3,000 | 2,890 ± 40 | −3.6 | ||
| 10,000 | 9,460 ± 140 | −5.4 | ||
| June 6, 2011 | July 19, 2011 (feeder) | 0 | BLOQ | NA |
| 300 | 283 ± 3 | −5.7 | ||
| 1,000 | 973 ± 3 | −2.7 | ||
| 3,000 | 2,950 ± 10 | −1.7 | ||
| 10,000 | 9,780 ± 10 | −2.2 | ||
| July 19, 2011 (bucket) | 0 | BLOQ | NA | |
| 300 | 302 ± 1 | 0.5 | ||
| 1,000 | 1,000 ± 10 | 0.3 | ||
| 3,000 | 3,000 ± 30 | 0.1 | ||
| 10,000 | 9,770 ± 120 | −0.3 | ||
| January 12, 2012 | February 23, 2012 (feeder) | 0 | BLOQ | NA |
| 300 | 299 ± 4 | −0.2 | ||
| 1,000 | 925 ± 1 | −7.5 | ||
| 3,000 | 2,790 ± 30 | −6.9 | ||
| 10,000 | 9,260 ± 110 | −7.4 | ||
| February 23, 2012 (bucket) | 0 | BLOQ | NA | |
| 300 | 297 ± 3 | −0.9 | ||
| 1,000 | 980 ± 6 | −2.0 | ||
| 3,000 | 2,900 ± 30 | −3.4 | ||
| 10,000 | 9,360 ± 130 | −6.4 | ||
| July 30, 2012 | September 5, 2012 (feeder) | 0 | BLOQ | NA |
| 300 | 282 ± 4 | −6.1 | ||
| 1,000 | 877 ± 35 | −12.3 | ||
| 3,000 | 2,850 ± 10 | −5.1 | ||
| 10,000 | 9,200 ± 250 | −8.0 | ||
| September 5, 2012 (bucket) | 0 | BLOQ | NA | |
| 300 | 288 ± 2 | −4.1 | ||
| 1,000 | 1,000 ± 80 | 0.0 | ||
| 3,000 | 2,830 ± 130 | −5.7 | ||
| 10,000 | 9,920 ± 280 | −0.8 | ||
Figure A-1. Reference Infrared Absorption Spectrum of Di(2-ethylhexyl) Phthalate
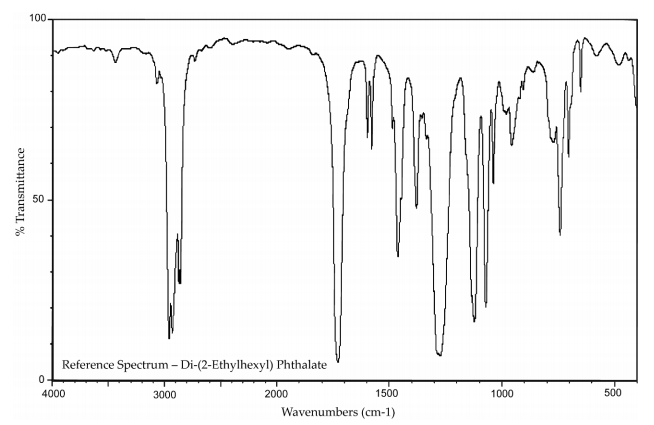
Reference spectra: National Institute of Advanced Industrial Science and Technology Spectral Database for Organic Compounds. SDBS No. 2266, https://sdbs.db.aist.go.jp/sdbs/cgi-bin/landingpage?spcode=IR-NIDA-08692 (accessed Mar 19, 2009).
Figure A-2. Fourier Transformed 1H Nuclear Magnetic Resonance Spectrum of Sample of Di(2-ethylhexyl) Phthalate (Lot 01514TH)
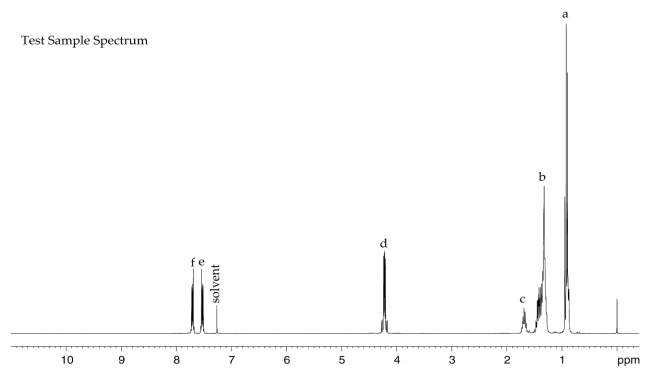
Reference spectra: National Institute of Advanced Industrial Science and Technology Spectral Database for Organic Compounds. SDBS No. 2266, https://sdbs.db.aist.go.jp/sdbs/cgi-bin/landingpage?spcode=NMR-HSP-03-645 (accessed Sep 22, 2009).
Figure A-3. Fourier Transformed 13C Nuclear Magnetic Resonance Spectrum of Sample of Di(2-ethylhexyl) Phthalate (Lot 01514TH)
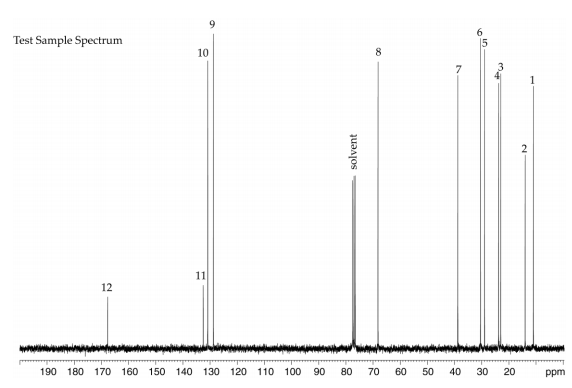
Reference spectra: National Institute of Advanced Industrial Science and Technology Spectral Database for Organic Compounds. SDBS No. 2266, https://sdbs.db.aist.go.jp/sdbs/cgi-bin/landingpage?spcode=NMR-CDS-05-148 (accessed Sep 22, 2009).
Appendix B. Ingredients, Nutrient Composition, and Contaminant Levels in NIH-07 and NTP-2000 Rat Rations
B.1.
NIH-07 Feed
Table B-1. Ingredients of NIH-07 Rat Ration
| Ingredients | Percent by Weight |
|---|---|
| Ground hard winter wheat | 23.00 |
| Ground #2 yellow shelled corn | 24.25 |
| Wheat middlings | 10.0 |
| Oat hulls | 0.0 |
| Alfalfa meal (dehydrated, 17% protein) | 4.0 |
| Purified cellulose | 0.0 |
| Soybean meal (49% protein) | 12.0 |
| Fish meal (60% protein) | 10.0 |
| Corn oil (without preservatives) | 0.0 |
| Soy oil (without preservatives) | 2.5 |
| Dried brewer’s yeast | 2.0 |
| Calcium carbonate (USP) | 0.5 |
| Vitamin premixa | 0.25 |
| Mineral premixb | 0.15 |
| Calcium phosphate, dibasic (USP) | 1.25 |
| Sodium chloride | 0.5 |
| Choline chloride (70% choline) | 0.10 |
| Dried skim milk | 5.00 |
| Dried molasses | 1.50 |
| Corn gluten meal (60% protein) | 3.00 |
| Methionine | 0.0 |
Table B-2. Vitamins and Minerals in NIH-07 Rat Ration
| Amounta | Source | |
|---|---|---|
| Vitamins | ||
| Vitamin A | 6,062 IU | Stabilized vitamin A palmitate or acetate |
| Vitamin D | 5,070 IU | D-activated animal sterol |
| Vitamin K | 3.1 mg | Menadione sodium bisulfite complex |
| Vitamin E | 22 IU | α-Tocopheryl acetate |
| Niacin | 33 mg | – |
| Folic acid | 2.4 mg | – |
| d-Pantothenic acid | 19.8 mg | d-Calcium pantothenate |
| Riboflavin | 3.8 mg | – |
| Thiamine | 11 mg | Thiamine mononitrate |
| B12 | 50 µg | – |
| Pyridoxine | 6.5 mg | Pyridoxine hydrochloride |
| Biotin | 0.15 mg | d-Biotin |
| Minerals | ||
| Iron | 132 mg | Iron sulfate |
| Zinc | 18 mg | Zinc oxide |
| Manganese | 66 mg | Manganese oxide |
| Copper | 4.4 mg | Copper sulfate |
| Iodine | 2.0 mg | Calcium iodate |
| Cobalt | 0.44 mg | Cobalt carbonate |
Table B-3. Nutrient Composition of NIH-07 Rat Ration
| Nutrient | Mean ± Standard Deviation | Range | Number of Samples |
|---|---|---|---|
| Protein (% by weight) | 23.65 ± 0.070 | 23.6–23.7 | 2 |
| Crude fat (% by weight) | 5.15 ± 0.212 | 5.0–5.3 | 2 |
| Crude fiber (% by weight) | 3.29 ± 0.042 | 3.26–3.32 | 2 |
| Ash (% by weight) | 6.015 ± 0.092 | 5.95–6.08 | 2 |
| Amino acids (% of total diet) | |||
| Arginine | 1.380 ± 0.06 | 1.3–1.49 | 10 |
| Cystine | 0.322 ± 0.031 | 0.274–0.372 | 10 |
| Glycine | 1.150 ± 0.070 | 1.06–1.31 | 10 |
| Histidine | 0.518 ± 0.024 | 0.497–0.553 | 10 |
| Isoleucine | 0.984 ± 0.024 | 0.952–1.03 | 10 |
| Leucine | 2.018 ± 0.067 | 1.93–2.13 | 10 |
| Lysine | 1.243 ± 0.051 | 1.13–1.32 | 10 |
| Methionine | 0.488 ± 0.016 | 0.468–0.515 | 10 |
| Phenylalanine | 1.097 ± 0.022 | 1.07–1.12 | 10 |
| Threonine | 0.918 ± 0.031 | 0.883–0.961 | 10 |
| Tryptophan | 0.277 ± 0.020 | 0.265–0.326 | 10 |
| Tyrosine | 0.860 ± 0.037 | 0.785–0.894 | 10 |
| Valine | 1.134 ± 0.025 | 1.11–1.17 | 10 |
| Essential fatty acids (% of total diet) | |||
| Linoleic | 2.30 ± 0.219 | 1.99–2.59 | 10 |
| Linolenic | 0.25 ± 0.275 | 0.217–0.296 | 10 |
| Vitamins | |||
| Vitamin A (IU/kg) | 6,020 ± 65.05 | 5,560–6,480 | 2 |
| α-Tocopherol (ppm) | 6,704 ± 21,045 | 40.3–66,600 | 10 |
| Thiamine (ppm)a | 14.2 ± 0.566 | 13.8–14.6 | 2 |
| Riboflavin (ppm) | 14.47 ± 3.352 | 10.0–19.8 | 10 |
| Niacin (ppm) | 99.33 ± 8.235 | 87.0–112.0 | 10 |
| Pantothenic acid (ppm) | 44.38 ± 3.806 | 38.2–51.1 | 10 |
| Pyridoxine (ppm)a | 12.876 ± 3.171 | 9.63–19.7 | 10 |
| Folic acid (ppm) | 2.482 ± 0.487 | 1.68–3.09 | 10 |
| Biotin (ppm) | 0.3283 ± 0.172 | 0.0–0.638 | 10 |
| B12 (ppb) | 49.4 ± 6.83 | 41.8–61.6 | 10 |
| Choline (as chloride) (ppm) | 1,821.0 ± 197.5 | 1,570–2,200 | 10 |
| Minerals | |||
| Calcium (%) | 1.004 ± 0.008 | 0.998–1.01 | 2 |
| Phosphorus (%) | 0.910 ± 0.002 | 0.908–0.911 | 2 |
| Potassium (%) | 0.830 ± 0.036 | 0.769–0.88 | 10 |
| Chloride (%) | 0.652 ± 0.106 | 0.441–0.8 | 10 |
| Sodium (%) | 0.378 ± 0.46 | 0.318–0.469 | 10 |
| Magnesium (%) | 0.187 ± 0.014 | 0.17–0.218 | 10 |
| Iron (ppm) | 385.1 ± 54.9 | 276.0–469.0 | 10 |
| Manganese (ppm) | 90.81 ± 7.566 | 80.7–104.0 | 10 |
| Zinc (ppm) | 64.15 ± 10.07 | 52.4–89.2 | 10 |
| Copper (ppm) | 14.13 ± 2.57 | 11.9–21.1 | 10 |
| Iodine (ppm) | 1.811 ± 0.992 | 0.54–3.45 | 10 |
| Chromium (ppm) | 3.946 ± 0.036 | 3.89–4.0 | 8 |
| Cobalt (ppm) | 0.5155 ± 0.267 | 0.01–0.963 | 10 |
Table B-4. Contaminant Levels in NIH-07 Rat Ration
| Mean ± Standard Deviation | Range | Number of Samples | |
|---|---|---|---|
| Contaminants | |||
| Arsenic (ppm) | 0.3865 ± 0.013 | 0.377–0.396 | 2 |
| Cadmium (ppm) | 0.0875 ± 0.004 | 0.085–0.09 | 2 |
| Lead (ppm) | 0.072 ± 0.004 | 0.069–0.074 | 2 |
| Mercury (ppm) | 0.013 ± 0.001 | 0.012–0.014 | 2 |
| Selenium (ppm) | 0.382 ± 0.014 | 0.372–0.392 | 2 |
| Aflatoxins (ppb)a | 5 | – | 2 |
| Nitrate nitrogen (ppm)b | 11.8 ± 2.55 | 10.0–13.6 | 2 |
| Nitrite nitrogen (ppm)a,b | <0.61 | – | 2 |
| BHA (ppm)a,c | <1.0 | – | 2 |
| BHT (ppm)a,c | <1.0 | – | 2 |
| Aerobic plate count (CFU/gm) | 60 ± 70.7 | 10–110 | 2 |
| Coliform (MPN/gm) | <3.0 | – | 2 |
| Escherichia coli (MPN/gm) | <10 | – | 2 |
| Salmonella (MPN/gm) | Negative | – | 2 |
| Total nitrosamines (ppb)d | 5.5 ± 1.768 | 4.2–6.7 | 2 |
| N-nitrosodimethylamine (ppb)d | 4.5 ± 1.768 | 3.2–5.7 | 2 |
| N-nitrosopyrrolidine (ppb)d | 1 ± 0.0 | 1.0–1.0 | 2 |
| Pesticides (ppm) | |||
| α-BHCa | <0.01 | – | 2 |
| β-BHCa | <0.02 | – | 2 |
| γ-BHCa | <0.01 | – | 2 |
| δ-BHCa | <0.01 | – | 2 |
| Heptachlora | <0.01 | – | 2 |
| Aldrina | <0.01 | – | 2 |
| Heptachlor epoxidea | <0.01 | – | 2 |
| DDEa | <0.01 | – | 2 |
| DDDa | <0.01 | – | 2 |
| DDTa | <0.01 | – | 2 |
| HCBa | <0.01 | – | 2 |
| Mirexa | <0.01 | – | 2 |
| Methoxychlora | <0.05 | – | 2 |
| Dieldrina | <0.01 | – | 2 |
| Endrina | <0.01 | – | 2 |
| Telodrina | <0.01 | – | 2 |
| Chlordanea | <0.05 | – | 2 |
| Toxaphenea | <0.10 | – | 2 |
| Estimated PCBsa | <0.20 | – | 2 |
| Ronnela | <0.01 | – | 2 |
| Ethiona | <0.02 | – | 2 |
| Trithiona | <0.05 | – | 2 |
| Diazinona | <0.10 | – | 2 |
| Methyl chlorpyrifos | <0.02 | – | 2 |
| Methyl parathiona | <0.02 | – | 2 |
| Ethyl parathiona | <0.02 | – | 2 |
| Malathion | 0.081 ± 0.082 | 0.024–0.139 | 2 |
| Endosulfan Ia | <0.01 | – | 2 |
| Endosulfan IIa | <0.01 | – | 2 |
| Endosulfane sulfatea | <0.03 | – | 2 |
B.2.
NTP-2000 Feed
Table B-5. Ingredients of NTP-2000 Rat Ration
| Ingredients | Percent by Weight |
|---|---|
| Ground hard winter wheat | 22.26 |
| Ground #2 yellow shelled corn | 22.18 |
| Wheat middlings | 15.0 |
| Oat hulls | 8.5 |
| Alfalfa meal (dehydrated, 17% protein) | 7.5 |
| Purified cellulose | 5.5 |
| Soybean meal (49% protein) | 5.0 |
| Fish meal (60% protein) | 4.0 |
| Corn oil (without preservatives) | 3.0 |
| Soy oil (without preservatives) | 3.0 |
| Dried brewer’s yeast | 1.0 |
| Calcium carbonate (USP) | 0.9 |
| Vitamin premixa | 0.5 |
| Mineral premixb | 0.5 |
| Calcium phosphate, dibasic (USP) | 0.4 |
| Sodium chloride | 0.3 |
| Choline chloride (70% choline) | 0.26 |
| Methionine | 0.2 |
Table B-6. Vitamins and Minerals in NTP-2000 Rat Ration
| Amounta | Source | |
|---|---|---|
| Vitamins | ||
| Vitamin A | 4,000 IU | Stabilized vitamin A palmitate or acetate |
| Vitamin D | 1,000 IU | D-activated animal sterol |
| Vitamin K | 1.0 mg | Menadione sodium bisulfite complex |
| α-Tocopheryl acetate | 100 IU | – |
| Niacin | 23 mg | – |
| Folic acid | 1.1 mg | – |
| d-Pantothenic acid | 10 mg | d-Calcium pantothenate |
| Riboflavin | 3.3 mg | – |
| Thiamine | 4 mg | Thiamine mononitrate |
| B12 | 52 µg | – |
| Pyridoxine | 6.3 mg | Pyridoxine hydrochloride |
| Biotin | 0.2 mg | d-Biotin |
| Minerals | ||
| Magnesium | 514 mg | Magnesium oxide |
| Iron | 35 mg | Iron sulfate |
| Zinc | 12 mg | Zinc oxide |
| Manganese | 10 mg | Manganese oxide |
| Copper | 2.0 mg | Copper sulfate |
| Iodine | 0.2 mg | Calcium iodate |
| Chromium | 0.2 mg | Chromium acetate |
Table B-7. Nutrient Composition of NTP-2000 Rat Ration
| Nutrient | Mean ± Standard Deviation | Range | Number of Samples |
|---|---|---|---|
| Protein (% by weight) | 14.78 ± 0.543 | 13.9–16.8 | 28 |
| Crude fat (% by weight) | 8.63 ± 0.387 | 8.0–9.7 | 28 |
| Crude fiber (% by weight) | 9.37 ± 0.534 | 7.49–10.1 | 28 |
| Ash (% by weight) | 5.23 ± 1.767 | 4.6–14.2 | 28 |
| Amino acids (% of total diet) | |||
| Arginine | 0.805 ± 0.075 | 0.67–0.97 | 29 |
| Cystine | 0.220 ± 0.021 | 0.15–0.25 | 29 |
| Glycine | 0.702 ± 0.038 | 0.62–0.80 | 29 |
| Histidine | 0.342 ± 0.070 | 0.27–0.68 | 29 |
| Isoleucine | 0.549 ± 0.040 | 0.43–0.66 | 29 |
| Leucine | 1.100 ± 0.063 | 0.96–1.24 | 29 |
| Lysine | 0.700 ± 0.104 | 0.31–0.86 | 29 |
| Methionine | 0.409 ± 0.042 | 0.26–0.49 | 29 |
| Phenylalanine | 0.623 ± 0.047 | 0.471–0.72 | 29 |
| Threonine | 0.513 ± 0.041 | 0.43–0.61 | 29 |
| Tryptophan | 0.155 ± 0.027 | 0.11–0.2 | 29 |
| Tyrosine | 0.422 ± 0.066 | 0.28–0.54 | 29 |
| Valine | 0.666 ± 0.040 | 0.55–0.73 | 29 |
| Essential fatty acids (% of total diet) | |||
| Linoleic | 3.94 ± 0.235 | 3.49–4.55 | 29 |
| Linolenic | 0.30 ± 0.064 | 0.005–0.368 | 29 |
| Vitamins | |||
| Vitamin A (IU/kg) | 3,886 ± 81.3 | 2,030–5,290 | 28 |
| α-Tocopherol (ppm) | 2,456 ± 12,817 | 13.6–69,100 | 29 |
| Thiamine (ppm)a | 7.96 ± 0.484 | 3.9–11.1 | 28 |
| Riboflavin (ppm) | 8.17 ± 2.841 | 4.2–17.5 | 29 |
| Niacin (ppm) | 78.66 ± 8.11 | 66.4–98.2 | 29 |
| Pantothenic acid (ppm) | 26.42 ± 11.05 | 17.4–81.0 | 29 |
| Pyridoxine (ppm)a | 9.75 ± 2.045 | 6.44–14.3 | 29 |
| Folic acid (ppm) | 1.58 ± 0.43 | 1.15–3.27 | 29 |
| Biotin (ppm) | 0.323 ± 0.093 | 0.2–0.704 | 29 |
| B12 (ppb) | 50.41 ± 34.89 | 18.3–174 | 29 |
| Choline (as chloride) (ppm) | 2,593 ± 633.8 | 1,160–3,790 | 29 |
| Minerals | |||
| Calcium (%) | 0.905 ± 0.041 | 0.831–1.03 | 28 |
| Phosphorus (%) | 0.540 ± 0.097 | 0.053–0.60 | 28 |
| Potassium (%) | 0.668 ± 0.029 | 0.626–0.733 | 29 |
| Chloride (%) | 0.392 ± 0.044 | 0.3–0.517 | 29 |
| Sodium (%) | 0.195 ± 0.027 | 0.16–0.283 | 29 |
| Magnesium (%) | 0.217 ± 0.054 | 0.185–0.49 | 29 |
| Iron (ppm) | 191.6 ± 36.18 | 135–311 | 29 |
| Manganese (ppm) | 50.11 ± 9.42 | 21–73.1 | 29 |
| Zinc (ppm) | 57.3 ± 25.54 | 43.3–184 | 29 |
| Copper (ppm) | 7.57 ± 2.49 | 3.21–16.3 | 29 |
| Iodine (ppm) | 0.513 ± 0.221 | 0–0.972 | 29 |
| Chromium (ppm) | 1.02 ± 1.04 | 0.33–3.97 | 28 |
| Cobalt (ppm) | 0.222 ± 0.152 | 0.0857–0.864 | 27 |
Table B-8. Contaminant Levels in NTP-2000 Rat Ration
| Mean ± Standard Deviation | Range | Number of Samples | |
|---|---|---|---|
| Contaminants | |||
| Arsenic (ppm) | 0.2 ± 0.048 | 0.147–0.383 | 28 |
| Cadmium (ppm) | 0.051 ± 0.008 | 0.038–0.082 | 28 |
| Lead (ppm) | 0.144 ± 0.110 | 0.064–0.474 | 28 |
| Mercury (ppm) | 0.0115 ± 0.004 | 0.01–0.03 | 28 |
| Selenium (ppm) | 0.161 ± 0.034 | 0.029–0.242 | 28 |
| Aflatoxins (ppb)a | <5.0 | – | 28 |
| Nitrate nitrogen (ppm)b | 15.7 ± 5.98 | 10.0–35.1 | 28 |
| Nitrite nitrogen (ppm)a,b | <0.61 | – | 28 |
| BHA (ppm)a,c | <1.00 | – | 28 |
| BHT (ppm)a,c | <1.00 | – | 28 |
| Aerobic plate count (CFU/gm) | <10.0 | – | 28 |
| Coliform (MPN/gm) | <3 | – | 28 |
| Escherichia coli (MPN/gm) | <10.0 | – | 28 |
| Salmonella (MPN/gm) | Negative | – | 28 |
| Total nitrosamines (ppb)d | 10.5 ± 6.01 | 1.5–24.5 | 28 |
| N-Ndimethylamine (ppb)d | 2.2 ± 1.50 | 0–6.6 | 28 |
| N-Npyrrolidine (ppb)d | 8.3 ± 5.47 | 1.4–20.0 | 28 |
| Pesticides (ppm) | |||
| α-BHCa | <0.01 | – | 28 |
| β-BHCa | <0.02 | – | 28 |
| γ-BHCa | <0.01 | – | 28 |
| δ-BHCa | <0.01 | – | 28 |
| Heptachlora | <0.01 | – | 28 |
| Aldrina | <0.01 | – | 28 |
| Heptachlor epoxidea | <0.01 | – | 28 |
| DDEa | <0.01 | – | 28 |
| DDDa | <0.01 | – | 28 |
| DDTa | <0.01 | – | 28 |
| HCBa | <0.01 | – | 28 |
| Mirexa | <0.01 | – | 28 |
| Methoxychlora | <0.05 | – | 28 |
| Dieldrina | <0.01 | – | 28 |
| Endrina | <0.01 | – | 28 |
| Telodrina | <0.01 | – | 28 |
| Chlordanea | <0.05 | – | 28 |
| Toxaphenea | <0.10 | – | 28 |
| Estimated PCBsa | <0.20 | – | 28 |
| Ronnela | <0.01 | – | 28 |
| Ethiona | <0.02 | – | 28 |
| Trithiona | <0.05 | – | 28 |
| Diazinona | <0.10 | – | 28 |
| Methyl chlorpyrifos | 0.092 ± 0.075 | 0.2–0.315 | 28 |
| Methyl parathiona | <0.02 | – | 28 |
| Ethyl parathiona | <0.02 | – | 28 |
| Malathion | 0.071 ± 0.07 | 0.02–0.297 | 28 |
| Endosulfan Ia | <0.01 | – | 28 |
| Endosulfan IIa | <0.01 | – | 28 |
| Endosulfane sulfatea | <0.03 | – | 28 |
Appendix C. Sentinel Animal Program
C.1.
Methods
Rodents used in the National Toxicology Program are produced in optimally clean facilities to eliminate potential pathogens that might affect study results. The Sentinel Animal Program is part of the periodic monitoring of animal health that occurs during the toxicological evaluation of test compounds. Under this program, the disease state of the rodents is monitored via sera or feces from extra (sentinel) or exposed animals in the study rooms. The sentinel animals and the study animals are subject to identical environmental conditions. Furthermore, the sentinel animals are from the same production source and weanling groups as the animals used for the studies of test compounds.
For these toxicology and carcinogenesis studies, blood samples were collected from each sentinel animal, allowed to clot, and the serum was separated. Additionally, fecal samples were collected and tested for endoparasites and Helicobacter species. All samples were processed appropriately with serology and Helicobacter testing performed by IDEXX BioResearch (formerly Rodent Animal Diagnostic Laboratory [RADIL], University of Missouri), Columbia, MO, for determination of the presence of pathogens. Evaluation for endo- and ectoparasites was performed in-house by the testing laboratory.
The laboratory methods and agents for which testing was performed are tabulated in Table C-1, Table C-2 below; the times at which samples were collected during the studies are also listed.
C.2.
Results
C.2.1.
Perinatal and Postweaning Study (Study 1)
Rats: Positive for endoparasites, pinworms (Syphacia spp.). All other test results were negative.
C.2.2.
Postweaning-only Study (Study 2)
Rats: Positive for endoparasites, pinworms (Syphacia spp.). All other test results were negative.
Table C-1. Methods and Results for Sentinel Animal Testing in Male and Female Rats (Study 1)
| Collection Time Points | Quarantinea | 3.5 Weeksb | 4 Weeks Post-Study Startc | 6 Months | 12 Months | 18 Months | End of Study |
|---|---|---|---|---|---|---|---|
| Number examined (males/females) | 0/10 | 0/10 | 5/5 | 5/5 | 6/6d | 5/6e | 5/5 |
| Method/test | |||||||
| Multiplex fluorescent immunoassay (MFI) | |||||||
| Kilham rat virus (KRV) | − | − | − | − | − | − | − |
| Mycoplasma pulmonis | − | − | − | − | − | − | − |
| Parvo NS-1 | − | − | − | − | − | − | − |
| Pneumonia virus of mice (PVM) | − | − | − | − | − | − | − |
| Rat coronavirus/ sialodacryoadenitis virus (RCV/SDA) | − | − | − | − | − | − | − |
| Rat minute virus (RMV) | − | − | − | − | − | − | − |
| Rat parvo virus (RPV) | − | − | − | − | − | − | − |
| Rat theilovirus (RTV) | − | − | − | − | − | − | − |
| Sendai | − | − | − | − | − | − | − |
| Theiler’s murine encephalomyelitis virus (TMEV) | − | − | − | − | − | − | − |
| Toolan’s H-1 | − | − | − | − | − | − | − |
| Immunofluorescence assay (IFA) | |||||||
| Pneumocystis carinii | NT | NT | NT | − | NT | NT | NT |
| In-house evaluation | |||||||
| Endoparasite evaluation (evaluation of cecal content) | − | NT | NT | NT | + | + | NT |
| Ectoparasite evaluation (evaluation of perianal surface) | − | NT | NT | NT | − | − | NT |
Table C-2. Methods and Results for Sentinel Animal Testing in Male and Female Rats (Study 2)
| Collection Time Points | 4 Weeksa | 6 Months | 12 Months | 18 Months | End of Study |
|---|---|---|---|---|---|
| Number examined (males/females) | 5/5 | 5/5 | 5/5 | 5/5 | 5/5 |
| Method/test | |||||
| Multiplex fluorescent immunoassay (MFI) | |||||
| Kilham rat virus (KRV) | − | − | − | − | − |
| Mycoplasma pulmonis | − | − | − | − | − |
| Parvo NS-1 | − | − | − | − | − |
| Pneumonia virus of mice (PVM) | − | − | − | − | − |
| Rat coronavirus/sialodacryoadenitis virus (RCV/SDA) | − | − | − | − | − |
| Rat minute virus (RMV) | − | − | − | − | − |
| Rat parvo virus (RPV) | − | − | − | − | − |
| Rat theilovirus (RTV) | − | − | − | − | − |
| Sendai | − | − | − | − | − |
| Theiler’s murine encephalomyelitis virus (TMEV) | − | − | − | − | − |
| Toolan’s H-1 | − | − | − | − | − |
| Immunofluorescence assay (IFA) | |||||
| Kilham rat virus (KRV) | − | NT | NT | NT | NT |
| Pneumocystis carinii | NT | − | NT | NT | NT |
| Number examined (males/females) | 0/0 | 0/0 | 6/5 | 6/5 | 0/0 |
| Method/test | |||||
| In-house evaluation | |||||
| Endoparasite evaluation (evaluation of cecal content) | NT | NT | + | + | NT |
| Ectoparasite evaluation (evaluation of perianal surface) | NT | NT | + | + | NT |
Appendix D. Genetic Toxicology
D.1.
Rodent Chromosome Aberrations Test
D.1.1.
Methods
Female B6C3F1 mice (four animals per exposure group) were exposed to di(2-ethylhexyl) phthalate (DEHP) (0, 3,000, 6,000, or 12,000 ppm) in dosed feed for 14 days. The animals were subcutaneously implanted with a bromodeoxyuridine (BrdU) tablet241 18 hours before the scheduled harvest to allow selection of the appropriate cell population for scoring.242,243 Chromosomal aberrations induced by test article administration are present in maximum number at the first metaphase following exposure; they decline in number during subsequent nuclear divisions due to cell death. Two hours before sacrifice, the animals received an intraperitoneal injection of colchicine in saline. The animals were euthanized 18 hours after BrdU dosing. One or both femurs were removed, and the marrow was flushed out with phosphate-buffered saline (pH 7.0). Cells were treated with a hypotonic salt solution, fixed, and dropped onto chilled slides. After a 24-hour drying period, the slides were stained (with a modified fluorescence-plus-Giemsa technique) and scored.
Fifty first-division metaphase cells were scored from each animal. Responses were evaluated as the percentage of aberrant metaphase cells, excluding gaps. The data were analyzed by a trend test244 with p ≤ 0.025 considered to be significant. Pairwise comparisons of each exposure group to the corresponding solvent control group were considered significant for p ≤ 0.025/3 (the number of DEHP-exposed groups).
D.1.2.
Results
In vivo, no significant increases were observed in chromosomal aberrations in bone marrow cells of female B6C3F1 mice following administration of DEHP (3,000–12,000 ppm) in dosed feed for 14 days (Table D-1).
Table D-1. Chromosomal Aberrations in Mice Exposed to Di(2-ethylhexyl) Phthalate in Feed for 14 Daysa,b
| n | Percent Cells with Aberrations | P Value (Pairwise) | |
|---|---|---|---|
| DEHP (ppm) | |||
| 0 | 4 | 1.25 ± 0.48 | 0.0000 |
| 3,000 | 4 | 2.75 ± 1.11 | 0.0649 |
| 6,000 | 4 | 0.50 ± 0.29 | 0.8726 |
| 12,000 | 4 | 1.50 ± 0.87 | 0.3807 |
| Trendc | p = 0.6617 | ||
D.2.
In Vivo Micronucleus Test
D.2.1.
Methods
D.2.1.1.
Bone Marrow
Female B6C3F1 mice (10 animals per exposure group) were exposed to DEHP (0, 3,000, 6,000, or 12,000 ppm) in dosed feed for 14 days. Bone marrow smears were prepared from cells obtained from the femurs as described for the chromosomal aberrations assay. Air-dried smears were fixed and stained with acridine orange; 1,000 polychromatic erythrocytes (PCEs) were scored per animal for the frequency of micronucleated cells.
The results were tabulated as the mean of the pooled results from all animals within an exposure group ± the standard error of the mean. The frequency of micronucleated cells among PCEs was analyzed for positive trend over the four exposure groups using a one-tailed Cochran-Armitage trend test, followed by pairwise comparisons between each exposed group and the concurrent control group. In the presence of excess binomial variation, as detected by a binomial dispersion test, the binomial variance of the Cochran-Armitage test was adjusted upward in proportion to the excess variation. For a test to be considered positive, the trend test p value ≤0.025 is required, along with at least one significant exposure group (p ≤ 0.025 divided by the number of exposed groups). Ultimately, the final call is determined by scientific staff after considering the results of statistical analyses, the reproducibility of any effects observed, and the magnitudes of those effects.
D.2.1.2.
Peripheral Blood
A detailed discussion of this assay is presented by MacGregor et al.245 Two 26-week exposure protocols were used. In the first study, male and female TgAC (FVB/N) transgenic mice were exposed to DEHP (0, 1,500, 3,000, or 6,000 ppm) in dosed feed. In the second study, male and female TgAC (FVB/N) transgenic mice were exposed to DEHP (0, 100, 200, or 400 mg/kg) via dermal application. In both studies, at the end of the 26-week exposure period, peripheral blood samples were obtained. Smears were immediately prepared and fixed in absolute methanol. The methanol-fixed slides were stained with acridine orange and coded. Slides were scanned to determine the frequency of micronuclei in 1,000 normochromatic erythrocytes (NCEs) and 1,000 PCEs per animal.
The results were tabulated as the mean of the pooled results from all animals within an exposure group ± the standard error of the mean. The frequency of micronucleated NCEs was analyzed as described above for the bone marrow micronucleus test.
D.2.1.3.
Evaluation Protocol
These are the basic guidelines for arriving at an overall assay result for assays performed by the National Toxicology Program. Statistical as well as biological factors are considered. For an individual assay, the statistical procedures for data analysis have been described in the preceding protocols. There have been instances, however, in which multiple aliquots of a chemical were tested in the same assay, and differing results were obtained among aliquots and/or among laboratories. Results from more than one aliquot or from more than one laboratory are not simply combined into an overall result. Rather, all the data are critically evaluated, particularly with regard to pertinent protocol variations, in determining the weight of evidence for an overall conclusion of chemical activity in an assay. For in vitro assays conducted with and without exogenous metabolic activation, results from each testing condition are evaluated and reported separately. The summary table in the abstract of this Technical Report presents a result that represents a scientific judgement of the overall evidence for activity of the chemical in an assay.
D.2.2.
Results
DEHP was tested in three independent erythrocyte micronucleus tests and produced varying results (Table D-2, Table D-3, Table D-4). In an in vivo bone marrow micronucleus test, B6C3F1 female mice were exposed to DEHP (3,000–12,000 ppm) in dosed feed for 14 days; results were judged to be equivocal (Table D-2). In a second test, DEHP (1,500–6,000 ppm) induced an equivocal response in male TgAC (FVB/N) mice and a positive response in female TgAC (FVB/N) mice following exposure via dosed feed for 26 weeks (Table D-3). Another 26-week exposure test in TgAC (FVB/N) mice using dermal application of DEHP (100–400 mg/kg) produced negative results in both male and female mice (Table D-4).
Table D-2. Frequency of Micronuclei in the Bone Marrow of Female B6C3F1 Mice Exposed to Di(2-ethylhexyl) Phthalate in Feed for 14 Days
| Micronucleated PCEs/1,000 PCEsa | P Valueb | Micronucleated NCEs/1,000 NCEsa | P Valueb | |
|---|---|---|---|---|
| n | 10 | 10 | 10 | 10 |
| DEHP (ppm) | ||||
| 0 | 1.90 ± 0.43 | 0.70 ± 0.26 | ||
| 3,000 | 1.20 ± 0.33 | 0.8958 | 0.90 ± 0.31 | 0.3085 |
| 6,000 | 1.10 ± 0.38 | 0.9281 | 1.50 ± 0.37 | 0.0440 |
| 12,000 | 1.50 ± 0.34 | 0.7538 | 2.00 ± 0.42 | 0.0061 |
| Trendc | p = 0.6970 | p = 0.0020 | ||
Table D-3. Frequency of Micronuclei in Peripheral Blood Erythrocytes of Male and Female TgAC (FVB/N) Mice Exposed to Di(2-ethylhexyl) Phthalate in Feed for 26 Weeks
| Micronucleated PCEs/1,000 PCEsa | n | P Valueb | Micronucleated NCEs/1,000 NCEsa | n | P Valueb | PCEs (%)a | |
|---|---|---|---|---|---|---|---|
| Male | |||||||
| DEHP (ppm) | |||||||
| 0 | 2.00 ± 0.39 | 12 | 2.83 ± 0.44 | 12 | 3.46 ± 0.16 | ||
| 1,500 | – | – | 2.18 ± 0.40 | 11 | 0.7975 | – | |
| 3,000 | – | – | 3.00 ± 0.58 | 13 | 0.4182 | – | |
| 6,000 | 3.67 ± 0.76 | 9 | 0.0108 | 4.33 ± 1.17 | 9 | 0.0610 | 3.10 ± 0.14 |
| Trendc | p = 0.0110 | p = 0.0260 | |||||
| Female | |||||||
| DEHP (ppm) | |||||||
| 0 | 2.50 ± 0.56 | 10 | 1.40 ± 0.27 | 10 | 3.47 ± 0.63 | ||
| 1,500 | – | – | 2.31 ± 0.38 | 13 | 0.0592 | – | |
| 3,000 | – | – | 1.50 ± 0.43 | 6 | 0.4358 | – | |
| 6,000 | 1.27 ± 0.33 | 11 | 0.9804 | 3.27 ± 0.51 | 11 | 0.0027 | 3.18 ± 0.18 |
| Trendc | p = 0.9800 | p = 0.0040 | |||||
Table D-4. Frequency of Micronuclei in Peripheral Blood Erythrocytes of Male and Female TgAC (FVB/N) Mice Following Dermal Application of Di(2-ethylhexyl) Phthalate for 26 Weeks
| Micronucleated PCEs/1,000 PCEsa | n | P Valueb | Micronucleated NCEs/1,000 NCEsa | n | P Valueb | PCEs (%)a | |
|---|---|---|---|---|---|---|---|
| Male | |||||||
| DEHP (mg/kg) | |||||||
| 0 | 2.45 ± 0.51 | 11 | 3.45 ± 0.59 | 11 | 4.20 ± 0.34 | ||
| 100 | – | – | 3.17 ± 0.34 | 12 | 0.6480 | – | |
| 200 | – | – | 3.36 ± 0.37 | 14 | 0.5522 | – | |
| 400 | 3.86 ± 0.67 | 7 | 0.0467 | 2.71 ± 0.68 | 7 | 0.8056 | 3.21 ± 0.28 |
| Trendc | p = 0.0470 | p = 0.7700 | |||||
| Female | |||||||
| DEHP (mg/kg) | |||||||
| 0 | 2.55 ± 0.47 | 11 | 1.64 ± 0.36 | 11 | 4.86 ± 0.34 | ||
| 100 | – | – | 1.91 ± 0.41 | 11 | 0.3153 | – | |
| 200 | – | – | 2.36 ± 0.49 | 11 | 0.1137 | – | |
| 400 | 4.09 ± 0.56 | 11 | 0.0231 | 2.55 ± 0.45 | 11 | 0.0700 | 4.24 ± 0.19 |
| Trendc | p = 0.0230 | p = 0.0590 | |||||
Appendix E. Mono(2-ethylhexyl) Phthalate Internal Dose Assessment
E.1.
Sample Collection
Select dams and their litters were removed on gestation day (GD) 18 to quantify mono(2-ethylhexyl) phthalate (MEHP) plasma and tissue concentrations. On GD 18, blood was collected from the retroorbital sinus of randomly selected dams (n = 5 per exposure group). Blood samples were collected in tubes containing K3 EDTA (tripotassium ethylene diamine tetraacetic acid), centrifuged, and the plasma harvested. Amniotic fluid was collected and pooled by dam, and each dam’s fetuses were collected and pooled by litter. All samples were flash frozen in liquid nitrogen and stored frozen at approximately −20°C before shipment to RTI International (Research Triangle Park, NC) for analysis.
E.2.
Sample Analysis
MEHP, a metabolite of di(2-ethylhexyl) phthalate, was measured in dam plasma, amniotic fluid, and fetal homogenate using a validated analytical method. The analyte stability in each matrix was also confirmed; corresponding data are given in Table E-1.
MEHP stock solutions were prepared in methanol at 0.5 mg/mL and diluted in water to prepare working standard solutions. A working internal standard (deuterated MEHP ([2H4]MEHP), C/D/N Isotopes Inc., Pointe-Claire, Canada) solution was prepared similarly at 1 µg/mL.
Plasma calibration standards were prepared at seven concentrations (25 to 5,000 ng/mL) by spiking 25 µL of plasma with an appropriate concentration of working MEHP standards. Plasma, amniotic fluid, and fetal homogenate quality control (QC) standards were prepared similarly at 100 and 2,500 ng/mL. Fetal homogenates were prepared by homogenizing fetuses in deionized water (1 g fetus in 3 mL water, equivalent to 250 mg/mL homogenate). Matrix blanks, method blanks, and study samples were prepared similarly to the matrix standards above but using 25 µL of water in place of the MEHP working standard solution. To all samples, 25 μL of internal standard solution and 425 µL acetonitrile were added, vortexed, and centrifuged at approximately 8,000 g for 10 minutes at 4°C. The supernatant was transferred to clean vials for analysis. Study samples with responses greater than the highest calibration standard were diluted with corresponding extracted blank matrix to the validated range prior to analysis.
E.3.
Instrumentation and Quantitation
All samples were analyzed by ultra-performance liquid chromatography (UPLC) tandem mass spectrometry (MS/MS) using Waters ACQUITY UPLC (Milford, MA) coupled to an Applied Biosystems 4000 QTRAP (Sciex, Framingham, MA) mass spectrometer. Chromatography was performed using Waters ACQUITY UPLC BEH C18 column (2.1 × 50 mm, 1.8 μm). Mobile phases A (water) and B (acetonitrile) were run with a linear gradient from 20% B to 95% B in 4.5 minutes at a flow rate of 0.5 mL/minute. The electrospray ion source was operated in negative ion mode with a voltage of −4,500 volts and source temperature of 450°C. Transitions monitored for MEHP and [1H4] were 277.1 → 133.8 and 281.0 → 137.8, respectively.
Calibration curves relating the response ratio of analyte to internal standard and the concentration of MEHP in a matrix were constructed using a 1/X2 weighted linear regression. The concentrations of MEHP in the samples were calculated using response ratio, the regression equation, initial sample weight or volume, and dilution when applicable. The concentration was reported as ng/mL for plasma and amniotic fluid. The fetal homogenate concentration estimated in ng/mL of fetal homogenate was converted to ng/g fetus by using a conversion factor of 4 (i.e., 4 mL homogenate = 1 g of fetus).
The performance of the calibration curve was evaluated before the analysis of each sample set. A successful calibration was indicated by the following: correlation coefficient (r) ≥ 0.99; relative standard deviation (RSD) ≤ ± 15% (except at the limit of quantitation [LOQ] where RSD is ≤ ± 20%); relative error (RE) ≤ ± 15% (except at the LOQ where RE is ≤ ± 20%). Data from study samples were considered valid if they were bracketed by valid QC sets. In general, each sample set, method blanks, and controls were bracketed by two QC sets, which consisted of a calibration blank and two concentrations of calibration standards (QC low and QC high). A QC set passed when the measured concentration for QC standards was within 15% of its nominal value. If the QC standard failed, it was necessary to reanalyze the bracketed samples. Correlation coefficient, r, for all calibration curves was ≥0.99. All QC standards were within 15% of nominal concentrations for plasma, amniotic fluid, and fetal homogenates.
Low concentrations of MEHP were observed in plasma, amniotic fluid, and fetal homogenates used as blank matrices, some of which were attributed to background contributions from the reagents and vials used in the assay. The background contribution from the reagents/vials did not affect the quantitation of study samples because matrix calibration standards and study samples were prepared and quantified similarly. In general, the background concentrations estimated in amniotic fluid and fetal homogenates were slightly higher than those observed in plasma, although the reason is not clear.
Table E-1. Analytical Method Validation and Stability Data for Mono(2-ethylhexyl) Phthalate in Plasma, Amniotic Fluid, and Fetal Homogenatea
| Validation Parameter | Dam Plasma | Amniotic Fluid | Fetal Homogenate |
|---|---|---|---|
| Matrix concentration range (ng/mL) | 25–5,000 | – | – |
| LOQ (ng/mL or ng/g) | 25.0 | 50.0 | 200 |
| LODb (µg/g) | 5.2 | 12.0 | 10 |
| Correlation coefficient (r) | ≥0.998 | – | – |
| Selectivity (%)c | 40 | 53 | 54 |
| Recovery (%)d | 95.0–102.0 | – | – |
| Precision and accuracye,f | |||
| Intra-day % RSD | ≤4.1 | ≤4.2 | ≤7.1 |
| Intra-day % RE | ≤ ± 2.9 | ≤ ± 4.7 | ≤ ± 5.6 |
| Inter-day % RSD | ≤5.2 | – | – |
| Inter-day % RE | ≤ ± 5.8 | – | – |
| Dilution verification (50,000 ng/mL) | |||
| % RSD | 0.8 | – | – |
| % RE | 1.7 | – | – |
| Extracted sample storage stability (% of day 0)f | |||
| Ambient | 102–105 | – | – |
| Refrigerator | 97–107 | – | – |
| Freeze-thaw (3 cycles) | 103–106 | – | – |
| Matrix storage stability (% of day 0)g | 80–113 | 80–115 | 84–113 |
Appendix F. Benchmark Dose Analysis
F.1.
Methods
Benchmark doses (BMDs) were calculated using the EPA Benchmark Dose Software (BMDS), version 3.1.2.169 The dose variable for the models was the amount of di(2-ethylhexyl) phthalate (DEHP) consumed/kg body weight/day (mg/kg/day). Numbers of animals per exposure group were poly-3-adjusted survival numbers. The response variable was the incidence of the endpoint being modeled.
All of the frequentist dichotomous models in the BMDS were used. The logistic, log-probit, and probit models were used with no parameter restrictions. Other models (dichotomous Hill, gamma, log-logistic, multistage, and Weibull) were used with default restrictions on the ranges of some of the parameters, as described in the BMDS User Guide.170
The benchmark response (BMR) used in the models was 0.1 (10%) extra risk, with estimated background levels. The benchmark dose lower confidence limit (BMDL10) was calculated using a 95% confidence interval. The decision logic used to recommend one model from the fitted models was the default logic.170
F.2.
Results
F.2.1.
Hepatocellular Adenoma or Carcinoma (Combined; Male Rats)
F.2.1.1.
Perinatal and Postweaning Study
All models provided an adequate fit to the data as assessed by a chi-square goodness-of-fit test (p ≥ 0.1) and by visual inspection of the respective plots of observed versus predicted values from the various models (Table F-1; Figure F-1). The dichotomous Hill, logistic, and probit models provided similar fits to the data. The probit model was judged to provide the best model fit based on the lowest Akaike information criterion (AIC) value. The BMDS dichotomous results for the probit and other models are available as supplemental data (Appendix H).
F.2.1.2.
Postweaning-only Study
All models except for the dichotomous Hill and multistage degree 1 models provided an adequate fit to the data as assessed by a chi-square goodness-of-fit test (p ≥ 0.1) and by visual inspection of the respective plots of observed versus predicted values from the various models (Table F-1; Figure F-1). The multistage degree 4 and multistage degree 3 models provided similar fits to the data. The multistage degree 4 model was judged to provide the best model fit based on the lowest AIC value. The BMDS dichotomous results for the multistage degree 4 and other models are available as supplemental data (Appendix H).
F.2.2.
Hepatocellular Adenoma or Carcinoma (Combined; Female Rats)
F.2.2.1.
Perinatal and Postweaning Study
All models except for the logistic and probit models provided adequate fits of the data as assessed by a chi-square goodness-of-fit test (p ≥ 0.1) and by visual inspection of the respective plots of observed versus predicted values from the various models (Table F-2; Figure F-2). The log-logistic and log-probit models provided similar fits of the data. The log-logistic model was judged to provide the best model fit based on the lowest AIC value. The BMDS dichotomous results for the log-logistic and other models are available as supplemental data (Appendix H).
F.2.2.2.
Postweaning-only Study
All models provided adequate fits of the data as assessed by a chi-square goodness-of-fit test (p ≥ 0.1) and by visual inspection of the respective plots of observed versus predicted values from the various models (Table F-2; Figure F-2). The multistage degree 4, multistage degree 2, logistic, and probit models provided similar fits of the data. The multistage degree 4 model was judged to provide the best model fit based on the lowest AIC value. The BMDS dichotomous results for the multistage degree 4 and other models are available as supplemental data (Appendix H).
F.2.3.
Pancreatic Acinar Adenoma or Carcinoma (Combined; Male Rats)
F.2.3.1.
Perinatal and Postweaning Study
All models had poor goodness of fit (p < 0.1). In addition, all models other than dichotomous Hill, log-logistic, and log-probit had high residuals near the BMD10. Of the models without high residuals near the BMD10, the dichotomous Hill model had the best fit based on AIC and was the only model with chi-square p value >0.0001 (Table F-3; Figure F-3). The dichotomous Hill model provided the best model fit based on the highest chi-square p value and lowest AIC value. The BMDS dichotomous results for the dichotomous Hill model and other models are available as supplemental data (Appendix H).
F.2.3.2.
Postweaning-only Study
Only dichotomous Hill, log-logistic, and log-probit models provided adequate fits of the data as assessed by a chi-square goodness-of-fit test (p ≥ 0.1) and visual inspection of the respective plots of observed versus predicted values from the various models (Table F-3; Figure F-3). The dichotomous Hill, log-logistic, and log-probit models provided similar fits of the data. The log-logistic model provided the best model fit based on the highest chi-square p value and lowest AIC value. The BMDS dichotomous results for the log-logistic model and other models are available as supplemental data (Appendix H).
F.2.4.
Uterine (Including Cervix) Adenocarcinoma, Adenoma, Squamous Cell Carcinoma, or Squamous Cell Papilloma (Combined; Female Rats)
F.2.4.1.
Perinatal and Postweaning Study
All models provided adequate fits of the data as assessed by a chi-square goodness-of-fit test (p ≥ 0.1) and visual inspection of the respective plots of observed versus predicted values from the various models (Table F-4; Figure F-4). The multistage degree 1, logistic, and probit models provided similar fits of the data. The logistic model provided the best model fit based on the lowest AIC value. The BMDS dichotomous results for the logistic and other models are available as supplemental data (Appendix H).
F.2.4.2.
Postweaning-only Study
All models provided adequate fits of the data as assessed by a chi-square goodness-of-fit test (p ≥ 0.1) and visual inspection of the respective plots of observed versus predicted values from the various models (Table F-4; Figure F-4). The multistage degree 1, logistic, and probit models provided similar fits of the data. The probit model provided the best model fit based on the lowest AIC value. The BMDS dichotomous results for the probit and other models are available as supplemental data (Appendix H).
Table F-1. Benchmark Dose Modeling Results for Hepatocellular Adenoma or Carcinoma (Combined) in Male Rats in the Two-year Feed Studies of Di(2-ethylhexyl) Phthalate
| Model | Chi-square P Valuea |
AIC | BMD10 (mg/kg/day) |
BMDL10 (mg/kg/day) |
BMDS Recommendationb |
|---|---|---|---|---|---|
| Perinatal and postweaning study (Study 1) | |||||
| Dichotomous hill | 0.55 | 92.9 | 199.5 | 168.2 | Viable–alternate |
| Gamma | 0.48 | 93.4 | 328.0 | 199.7 | Viable–alternate |
| Log-logistic | 0.48 | 93.4 | 326.1 | 194.6 | Viable–alternate |
| Multistage degree 4 | 0.45 | 93.6 | 356.5 | 198.5 | Viable–alternate |
| Multistage degree 3 | 0.45 | 93.6 | 356.5 | 198.5 | Viable–alternate |
| Multistage degree 2 | 0.45 | 93.6 | 356.5 | 198.5 | Viable–alternate |
| Multistage degree 1 | 0.38 | 93.6 | 249.9 | 161.3 | Viable–alternate |
| Weibull | 0.47 | 93.5 | 336.2 | 198.8 | Viable–alternate |
| Logistic | 0.61 | 92.0 | 413.6 | 335.9 | Viable–alternate |
| Log-probit | 0.52 | 93.2 | 302.0 | 188.6 | Viable–alternate |
| Probit | 0.63 | 91.8 | 382.9 | 306.1 | Viable–recommended |
| Postweaning-only study (Study 2) | |||||
| Dichotomous hill | 0.04 | 91.6 | 223.1 | 177.2 | Questionable |
| Gamma | 0.14 | 89.6 | 416.2 | 262.1 | Viable–alternate |
| Log-logistic | 0.14 | 89.6 | 428.0 | 259.7 | Viable–alternate |
| Multistage degree 4 | 0.26 | 87.6 | 434.4 | 263.5 | Viable–recommended |
| Multistage degree 3 | 0.26 | 87.61 | 434.4 | 263.5 | Viable–alternate |
| Multistage degree 2 | 0.19 | 88.0 | 375.0 | 248.1 | Viable–alternate |
| Multistage degree 1 | 0.07 | 91.0 | 273.0 | 173.8 | Questionable |
| Weibull | 0.14 | 89.6 | 436.3 | 263.0 | Viable–alternate |
| Logistic | 0.18 | 87.9 | 421.5 | 349.6 | Viable–alternate |
| Log-probit | 0.13 | 89.6 | 393.9 | 250.8 | Viable–alternate |
| Probit | 0.15 | 88.1 | 395.2 | 321.4 | Viable–alternate |
Table F-2. Benchmark Dose Modeling Results for Hepatocellular Adenoma or Carcinoma (Combined) in Female Rats in the Two-year Feed Studies of Di(2-ethylhexyl) Phthalate
| Model | Chi-square P Valuea |
AIC | BMD10 (mg/kg/day) |
BMDL10 (mg/kg/day) |
BMDS Recommendationb | |
|---|---|---|---|---|---|---|
| Perinatal and postweaning study (Study 1) | ||||||
| Dichotomous hill | 0.14 | 143.1 | 77.6 | 41.5 | Viable–alternate | |
| Gamma | 0.14 | 142.7 | 153.7 | 105.9 | Viable–alternate | |
| Log-logistic | 0.24 | 141.5 | 122.9 | 79.7 | Viable–recommended | |
| Multistage degree 4 | 0.14 | 142.7 | 153.7 | 105.9 | Viable–alternate | |
| Multistage degree 3 | 0.14 | 142.7 | 153.7 | 105.9 | Viable–alternate | |
| Multistage degree 2 | 0.14 | 142.7 | 153.7 | 105.9 | Viable–alternate | |
| Multistage degree 1 | 0.14 | 142.7 | 153.7 | 105.9 | Viable–alternate | |
| Weibull | 0.14 | 142.7 | 153.7 | 105.9 | Viable–alternate | |
| Logistic | 0.02 | 148.2 | 348.5 | 274.2 | Questionable | |
| Log-probit | 0.27 | 142.1 | 92.0 | 41.5 | Viable–alternate | |
| Probit | 0.03 | 147.6 | 321.7 | 251.3 | Questionable | |
| Postweaning-only study (Study 2) | ||||||
| Dichotomous hill | 0.34 | 79.9 | 277.3 | 180.7 | Viable–alternate | |
| Gamma | 0.36 | 79.9 | 275.7 | 183.2 | Viable–alternate | |
| Log-logistic | 0.34 | 79.9 | 277.3 | 180.7 | Viable–alternate | |
| Multistage degree 4 | 0.81 | 77.0 | 383.6 | 208.0 | Viable–recommended | |
| Multistage degree 3 | 0.58 | 79.1 | 348.2 | 205.4 | Viable–alternate | |
| Multistage degree 2 | 0.69 | 77.5 | 302.0 | 197.2 | Viable–alternate | |
| Multistage degree 1 | 0.49 | 78.6 | 217.2 | 147.6 | Viable–alternate | |
| Weibull | 0.38 | 79.8 | 286.6 | 186.7 | Viable–alternate | |
| Logistic | 0.69 | 77.9 | 428.7 | 354.6 | Viable–alternate | |
| Log-probit | 0.36 | 80.2 | 338.5 | 168.0 | Viable–alternate | |
| Probit | 0.69 | 77.7 | 392.9 | 321.3 | Viable–alternate | |
Table F-3. Benchmark Dose Modeling Results for Pancreatic Acinar Adenoma or Carcinoma (Combined) in Male Rats in the Two-year Feed Studies of Di(2-ethylhexyl) Phthalate
| Model | Chi-square P Valuea |
AIC | BMD10 (mg/kg/day) |
BMDL10 (mg/kg/day) |
BMDS Recommendationb |
|---|---|---|---|---|---|
| Perinatal and postweaning study (Study 1) | |||||
| Dichotomous hill | 0.08 | 227.8 | 85.9 | 56.8 | Questionable |
| Gamma | <0.0001 | 257.0 | 52.9 | 37.9 | Questionable |
| Log-logistic | <0.0001 | 252.2 | 28.2 | 17.7 | Questionable |
| Multistage degree 4 | <0.0001 | 257.0 | 52.9 | 37.9 | Questionable |
| Multistage degree 3 | <0.0001 | 257.0 | 52.9 | 37.9 | Questionable |
| Multistage degree 2 | <0.0001 | 257.0 | 52.9 | 37.9 | Questionable |
| Multistage degree 1 | <0.0001 | 257.0 | 52.9 | 37.9 | Questionable |
| Weibull | <0.0001 | 257.0 | 52.9 | 37.9 | Questionable |
| Logistic | <0.0001 | 264.6 | 110.6 | 84.1 | Questionable |
| Log-probit | <0.0001 | 251.5 | 33.1 | 13.5 | Questionable |
| Probit | <0.0001 | 264.6 | 110.9 | 86.3 | Questionable |
| Postweaning-only study (Study 2) | |||||
| Dichotomous hill | 0.11 | 205.1 | 70.2 | 20.4 | Viable–alternate |
| Gamma | 0.07 | 205.3 | 44.7 | 35.0 | Questionable |
| Log-logistic | 0.13 | 204.6 | 31.0 | 20.2 | Viable–recommended |
| Multistage degree 4 | 0.07 | 205.3 | 44.7 | 35.0 | Questionable |
| Multistage degree 3 | 0.07 | 205.3 | 44.7 | 35.0 | Questionable |
| Multistage degree 2 | 0.07 | 205.3 | 44.7 | 35.0 | Questionable |
| Multistage degree 1 | 0.07 | 205.3 | 44.7 | 35.0 | Questionable |
| Weibull | 0.07 | 205.3 | 44.7 | 35.0 | Questionable |
| Logistic | 0.00 | 218.6 | 122.5 | 100.6 | Questionable |
| Log-probit | 0.11 | 204.9 | 32.6 | 15.3 | Viable–alternate |
| Probit | 0.00 | 217.6 | 115.8 | 96.9 | Questionable |
Table F-4. Benchmark Dose Modeling Results for Uterine (Including Cervix) Adenocarcinoma, Adenoma, Squamous Cell Carcinoma, or Squamous Cell Papilloma (Combined) in Female Rats in the Two-year Feed Studies of Di(2-ethylhexyl) Phthalate
| Model | Chi-square P Valuea |
AIC | BMD10 (mg/kg/day) |
BMDL10 (mg/kg/day) |
BMDS Recommendationb |
|---|---|---|---|---|---|
| Perinatal and postweaning study (Study 1) | |||||
| Dichotomous hill | 0.21 | 107.8 | 224.7 | 169.7 | Viable–alternate |
| Gamma | 0.42 | 106.0 | 560.0 | 289.2 | Viable–alternate |
| Log-logistic | 0.42 | 106.0 | 558.7 | 276.0 | Viable–alternate |
| Multistage degree 4 | 0.41 | 106.0 | 594.6 | 286.7 | Viable–alternate |
| Multistage degree 3 | 0.41 | 106.0 | 594.6 | 286.7 | Viable–alternate |
| Multistage degree 2 | 0.41 | 106.0 | 594.6 | 286.7 | Viable–alternate |
| Multistage degree 1 | 0.52 | 104.4 | 520.3 | 275.4 | Viable–alternate |
| Weibull | 0.42 | 106.0 | 566.6 | 288.7 | Viable–alternate |
| Logistic | 0.60 | 104.1 | 594.2 | 432.2 | Viable–recommended |
| Log-probit | 0.43 | 105.9 | 524.7 | 254.6 | Viable–alternate |
| Probit | 0.60 | 104.10 | 578.3 | 405.4 | Viable–alternate |
| Postweaning-only study (Study 2) | |||||
| Dichotomous hill | 0.33 | 147.6 | 180.6 | 116.6 | Viable–alternate |
| Gamma | 0.25 | 148.4 | 273.3 | 144.1 | Viable–alternate |
| Log-logistic | 0.25 | 148.4 | 267.5 | 127.5 | Viable–alternate |
| Multistage degree 4 | 0.25 | 148.5 | 272.6 | 143.1 | Viable–alternate |
| Multistage degree 3 | 0.25 | 148.5 | 272.9 | 143.1 | Viable–alternate |
| Multistage degree 2 | 0.25 | 148.5 | 272.6 | 143.1 | Viable–alternate |
| Multistage degree 1 | 0.41 | 146.6 | 224.4 | 141.7 | Viable–alternate |
| Weibull | 0.25 | 148.4 | 272.6 | 143.9 | Viable–alternate |
| Logistic | 0.39 | 146.7 | 344.0 | 268.9 | Viable–alternate |
| Log-probit | 0.26 | 148.2 | 260.3 | 96.7 | Viable–alternate |
| Probit | 0.41 | 146.6 | 324.1 | 249.0 | Viable–recommended |
Figure F-1. Benchmark Dose Modeling Results for Hepatocellular Adenoma or Carcinoma (Combined) in Male Rats in the Two-year Feed Studies of Di(2-ethylhexyl) Phthalate

BMR = benchmark response; BMD10 = benchmark dose corresponding to a 10% extra risk; BMDL10 = 95% lower bound on the benchmark dose corresponding to a 10% extra risk.
Figure F-2. Benchmark Dose Modeling Results for Hepatocellular Adenoma or Carcinoma (Combined) in Female Rats in the Two-year Feed Studies of Di(2-ethylhexyl) Phthalate
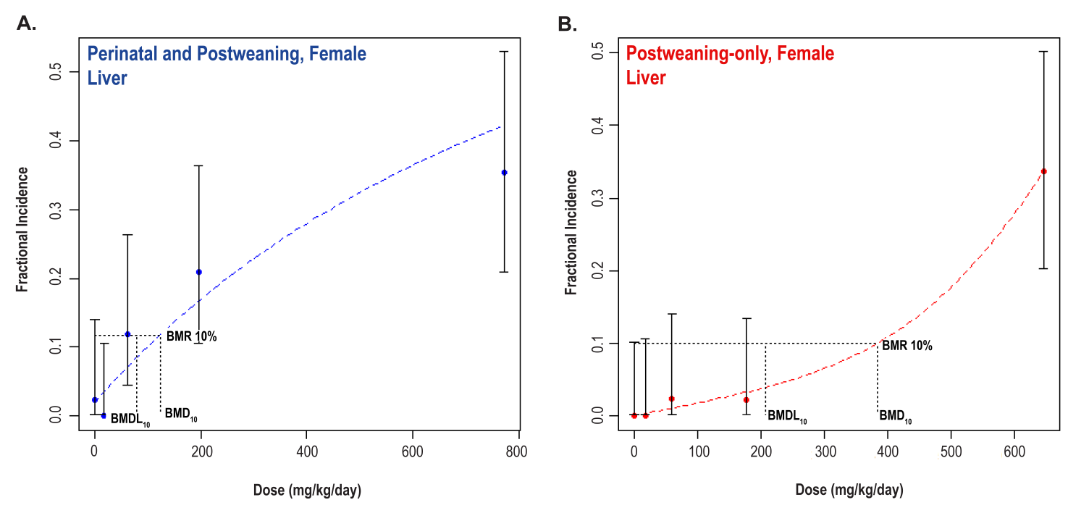
BMR = benchmark response; BMD10 = benchmark dose corresponding to a 10% extra risk; BMDL10 = 95% lower bound on the benchmark dose corresponding to a 10% extra risk.
Figure F-3. Benchmark Dose Modeling Results for Pancreatic Adenoma or Carcinoma (Combined) in Male Rats in the Two-year Feed Studies of Di(2-ethylhexyl) Phthalate
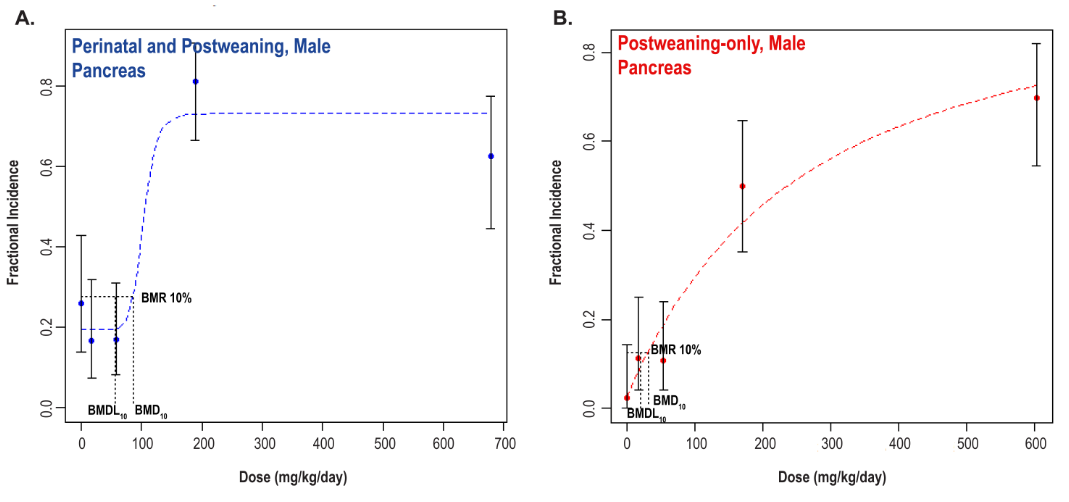
BMR = benchmark response; BMD10 = benchmark dose corresponding to a 10% extra risk; BMDL10 = 95% lower bound on the benchmark dose corresponding to a 10% extra risk.
Figure F-4. Benchmark Dose Modeling Results for Uterine (Including Cervix) Adenocarcinoma, Adenoma, Squamous Cell Carcinoma, or Squamous Cell Papilloma (Combined) in Female Rats in the Two-year Feed Studies of Di(2-ethylhexyl) Phthalate
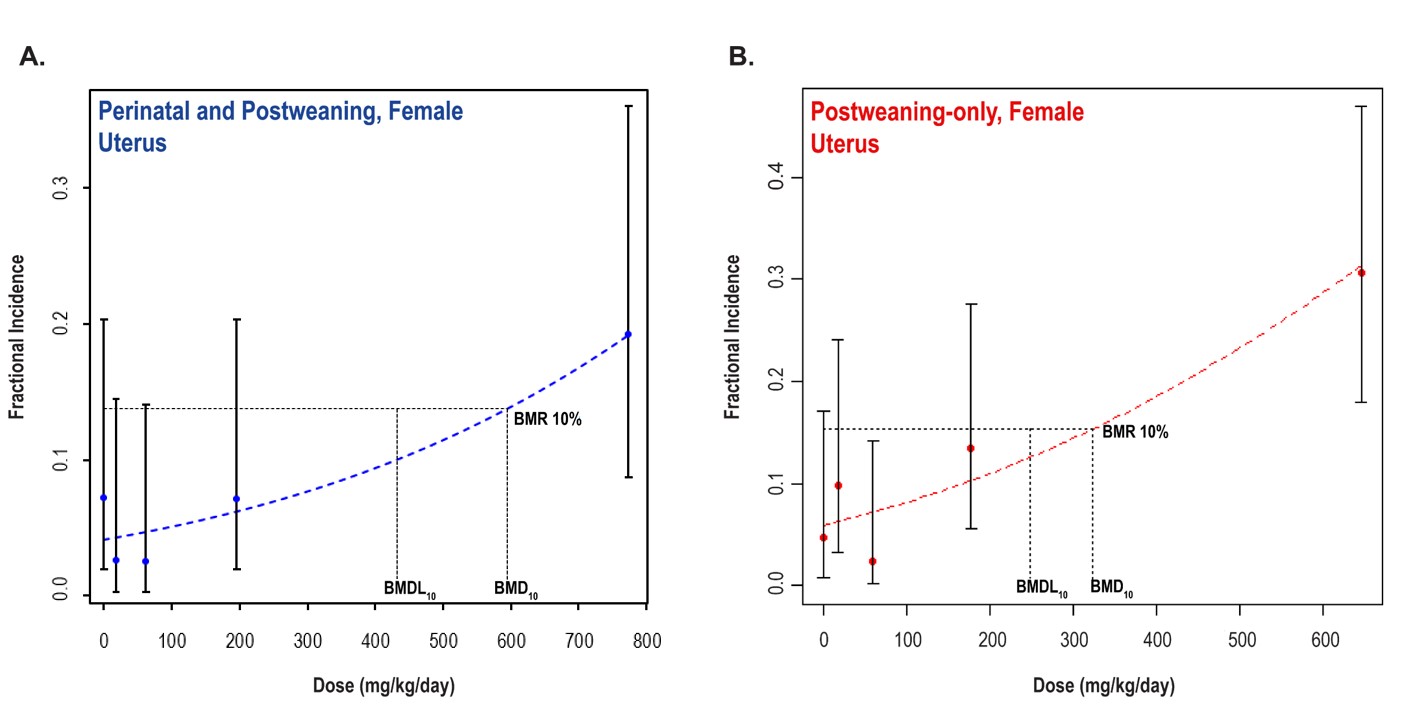
BMR = benchmark response; BMD10 = benchmark dose corresponding to a 10% extra risk; BMDL10 = 95% lower bound on the benchmark dose corresponding to a 10% extra risk.
Appendix G. Peer-review Report
The National Toxicology Program (NTP) virtually convened the NTP Technical Reports Peer-Review Panel (“the Panel”) on April 2, 2021, to peer review the Draft NTP Technical Reports on the Toxicology and Carcinogenesis Studies of Sodium Tungstate Dihydrate, Di-n-butyl Phthalate, and Di(2-ethylhexyl) Phthalate. Meeting information, including the draft reports, actions, and presentations is currently archived with NTP.
The panel peer reviewed the draft reports and provided its opinion on NTP’s preliminary conclusions regarding the level of evidence of carcinogenic activity of sodium tungstate dihydrate, di-n-butyl phthalate, and di(2-ethylhexyl) phthalate. The panel’s comments for the Draft NTP Technical Report on the Toxicology and Carcinogenesis Studies of Di(2-ethylhexyl) Phthalate (CASRN 117-81-7) Administered in Feed to Sprague Dawley (Hsd:Sprague Dawley SD) Rats begin at Section G.2.5. The panel’s recommendations do not necessarily represent NTP’s opinion.
G.1.
Attendees1
The meeting was held via webcast. Individuals who viewed the webcast are not listed except as noted.
Peer-Review Panel
Chair: Gabriele Ludewig, University of Iowa
Tracie E. Bunton, Eicarte, LLC
Michael R. Elwell, Apex Toxpath, LLC
Charles R. Mahrt, Retired, formerly with Flagship Biosciences
Daniel J. Spade, Brown University
John Pierce Wise, University of Louisville
National Toxicology Program Board of Scientific Counselors Liaison
Susan Felter, Procter & Gamble
National Institute of Environmental Health Sciences Staff
Mamta Behl
Chad Blystone
Mark Cesta
Sheba Churchill
Helen Cunny
Susan Elmore
Dori Germolec
Michelle Hooth
Madelyn (Mimi) Huang
Angela King-Herbert
Barry McIntyre
Georgia Roberts
Sheena Scruggs, Designated Federal Official
Kelly Shipkowski
Keith Shockley
Robert Sills
Stephanie Smith-Roe
Suramya Waidyanatha
Nigel Walker
Mary Wolfe
Other Federal Agency Staff
Shirisha Chittiboyina, National Institute for Occupational Safety and Health
Gonçalo Gamboa da Costa, U.S. Food and Drug Administration
Contract Support Staff
Amy Brix, EPL, Inc.
Canden Byrd, ICF
Lindsey Green, ICF
Shawn Harris, Social & Scientific Systems, a DLH Company
Elizabeth Maull, Kelly Government Services
Megan Rooney, ICF
Alessandria Schumacher, ICF
Cynthia Willson, Integrated Laboratory Systems, LLC
G.2.
Peer Review of the Draft NTP Technical Reports on the Toxicology and Carcinogenesis Studies of Sodium Tungstate Dihydrate, Di-n-butyl Phthalate, and Di(2-ethylhexyl) Phthalate
G.2.1.
Introductions and Welcome
The National Toxicology Program (NTP) convened a peer-review panel for the Draft NTP Technical Reports on the Toxicology and Carcinogenesis Studies of Sodium Tungstate Dihydrate, Di-n-butyl Phthalate, and Di(2-ethylhexyl) Phthalate on April 2, 2021 via webcast.
-
Dr. Gabriele Ludewig, panel chair, called the meeting to order at 10:00 a.m. and welcomed everyone to the meeting. She asked all attendees to introduce themselves, and reviewed the peer-review meeting format for the panel and audience.
-
Dr. Mary Wolfe, Acting Deputy Director for Policy & Communication, welcomed all participants to the meeting.
-
Dr. Sheena Scruggs read the conflict-of-interest policy statement and briefed the attendees on meeting logistics.
-
Dr. Susan Felter attended as the liaison to the NTP Board of Scientific Counselors.
-
Dr. Shirisha Chittiboyina attended as the liaison for the National Institute for Occupational Safety and Health.
-
Dr. Gonçalo Gamboa da Costa attended as the liaison for the U.S. Food and Drug Administration.
G.2.2.
Background and Charge to the Panel
Dr. Chad Blystone briefly presented the NTP draft technical report objectives, including a review of the levels of evidence for the potential carcinogenic activity and factors considered for tested chemicals. He also described how NTP collects historical control data2
on neoplastic lesions and how these are utilized to provide context to report findings. Dr. Blystone provided the charge for the individual peer reviews:
-
Review and evaluate the scientific and technical elements of the study and its presentation.
-
Determine whether the study’s experimental design, conduct, and findings support NTP’s conclusions under the conditions of this study.
The peer-review meeting materials can be found on the NTP website.
G.2.3.
Toxicology and Carcinogenesis Studies of Sodium Tungstate Dihydrate
G.2.3.1.
Presentation and Clarifying Questions
Dr. Mamta Behl summarized the studies and conclusions reported in the Draft NTP Technical Report on the Toxicology and Carcinogenesis Studies of Sodium Tungstate Dihydrate (CASRN 10213-10-2) in Sprague Dawley (Hsd:Sprague Dawley SD) Rats and B6C3F1/N Mice (Drinking Water Studies).
Tungsten occurs naturally in the environment and can enter waterways through the weathering of rocks and soil. It was nominated for study due to concerns about potential widespread human exposure via contaminated drinking water. Sodium tungstate dihydrate was selected because it is a naturally occurring, water-soluble form of tungsten. Drinking water was selected as the most likely route of exposure for the general population.
Dr. Behl first presented a summary of results from the perinatal and postweaning toxicity/carcinogenicity study in Hsd:Sprague Dawley SD rats. NTP exposed time-mated female rats to 0, 250, 500, or 1,000 mg/L sodium tungstate dihydrate in drinking water from gestational day (GD) 6 through postnatal day (PND) 21. NTP provided the F1 generation rats with the same respective sodium tungstate dihydrate concentrations as their dam for 2 years (n = 50/sex/concentration). In addition, F1 generation rats were provided dosed drinking water or the vehicle control for 3, 6, 12, or 18 months for interim evaluations (n = 40/sex/concentration).
Dr. Behl then presented a summary of results from the chronic toxicity/carcinogenicity study in B6C3F1/N mice. NTP exposed mice to 0, 500, 1,000, or 2,000 mg/L sodium tungstate dihydrate in drinking water for 2 years (n = 50/sex/concentration). An additional 40 mice/sex/concentration were included for interim evaluations at 3, 6, 12, and 18 months.
Under the conditions of these 2-year studies, NTP’s draft conclusions were:
-
No evidence of carcinogenic activity in male Hsd:Sprague Dawley SD rats at 250, 500, and 1,000 mg/L.
-
Exposure to sodium tungstate dihydrate in drinking water caused increased incidences of nonneoplastic lesions in the kidney of male rats.
-
Equivocal evidence of carcinogenic activity in female Hsd:Sprague Dawley SD rats based on increased incidences of C-cell adenoma or carcinoma (combined) of the thyroid gland.
-
Exposure to sodium tungstate dihydrate in drinking water caused increased incidences of nonneoplastic lesions in the kidney and uterus of female rats.
-
Equivocal evidence of carcinogenic activity in male B6C3F1/N mice based on the occurrences of renal tubule adenoma or carcinoma (combined) in exposed animals.
-
Exposure to sodium tungstate dihydrate in drinking water caused increased incidences of nonneoplastic lesions in the kidney, testes, and bone marrow of male mice.
-
No evidence of carcinogenic activity in female B6C3F1/N mice at 500, 1,000, and 2,000 mg/L sodium tungstate dihydrate.
-
Exposure to sodium tungstate dihydrate in drinking water caused increased incidences of nonneoplastic lesions in the kidney and spleen of female mice.
There were no clarifying questions or comments about the presentation.
G.2.3.2.
Public Comments
Dr. Ludewig acknowledged the receipt of one written public comment from Dr. Ranulfo Lemus on behalf of the International Tungsten Industry Association. Dr. Ludewig noted that the panel did not receive requests for oral public comments on the draft technical report.
G.2.3.3.
Peer-Review Comments and Panel Discussion
First Reviewer – Dr. Michael ElwellG.2.3.3.1
-
Dr. Elwell indicated that the dose selection was appropriate, the studies were well-conducted, the results were discussed clearly, and the rationale was clearly presented for neoplastic findings of equivocal evidence and several nonneoplastic effects. The important sodium tungstate-related findings were well-described and represented by quality pathology images in the report.
-
Dr. Elwell noted some inconsistencies across report sections on the relationship of atypical hyperplasia in the uterus of female rats to sodium tungstate dihydrate exposure. Text on page 104 of the draft report indicates that the relationship is unknown; however, text in the abstract, summary, and conclusions states that atypical hyperplasia in the uterus is a nonneoplastic effect caused by sodium tungstate dihydrate.
-
Dr. Amy Brix stated that the sentence in the discussion about the unknown relationship to atypical hyperplasia was a typo and NTP may consider removing it. She stated that NTP believes that the effects were related to exposure and would consider make those edits.
-
-
Dr. Elwell noted that on page 69 under “other tissues” for rats, several nonneoplastic findings were mentioned and considered to be of unknown biological significance. However, a significant decrease in fibroadenomas in the mammary gland was mentioned with no comment regarding biological significance or relationship to sodium tungstate dihydrate exposure. The fibroadenomas occurred in female rats with decreased body weights. He recommended that the report discuss the potential relevance of decreased body weight in relation to the tumors given that the effect of lower body weight on occurrence of this tumor has been noted in earlier NTP reports and in published studies.
-
Dr. Brix said that information about the mammary gland would be considered by NTP along with citing Dr. Haseman’s article that compares body weight changes to certain tumor incidences. For the nonneoplastic findings in other tissues, NTP can make it clear that they do not consider them toxicologically significant, treatment-related, or biologically significant.
-
-
Dr. Elwell questioned the rationale for including two of the effects listed in the abstract, specifically increased spleen hematopoiesis and bone marrow hyperplasia. For both findings, the increased incidences occurred in the low and mid-exposure groups with no significant effect in the high-exposure group, and the average severity was similar across exposure groups. Given that the histopathology section states that the biological significance for the bone marrow and spleen is unknown, Dr. Elwell asked if there were other effects (e.g., inflammatory, hematologic, or hematopoietic) that supported listing these as effects in the abstract? As a point of reference, Dr. Elwell noted that NTP concluded that the kidney pigment findings, which were significantly higher in the high-exposure group, were of questionable toxicologic importance and therefore not brought into the abstract.
-
Dr. Behl agreed with Dr. Elwell’s comment on the spleen and explained that NTP noted an effect in bone marrow hyperplasia in males and hematopoietic cell proliferation in females at both low and mid doses. NTP is open to discussion about whether to bring these effects forward into the abstract. Dr. Behl asked if the panel recommended including spleen effects in the abstract.
-
Dr. Elwell commented that the other effects in the abstract were dose- or target organ-related; the spleen effects were weaker than what might be expected for a finding listed in the abstract.
-
Dr. Brix noted that for the spleen and bone marrow, the incidences were two to three times higher in the low and mid-dose groups, but agreed that it is a weak connection and that NTP may be open to removing them from the abstract based on the panel discussion.
-
-
G.2.3.3.2
-
Dr. Wise concurred with Dr. Elwell. The report was well-written, and the study was well-designed.
-
Given the pressure to evaluate environmentally relevant concentrations, Dr. Wise recommended that NTP provide language on dose selection rationale to help readers unfamiliar with the NTP approach.
-
Dr. Behl noted that the comment on dose selection rationale was well-taken. She explained how NTP selected the concentrations and indicated that they have been criticized in the past for doses that failed to challenge the animals. The concentrations used in this study allowed NTP to state that tungstate at high levels does not result in overt toxicity. The effects observed in the kidney were common with this strain and species.
-
Dr. Wise clarified that it was not that NTP should use different doses. Rather, he recommended that NTP specify that their intention is to determine whether a substance causes cancer, not to define whether a substance causes cancer at the most environmentally relevant dose. It would help to add that context for a reader who does not understand that approach.
-
Dr. Behl said that NTP can add some clarifying language in the report.
-
-
-
Dr. Wise recommended that NTP provide clarifying language to address the occurrence of pinworms in the rats for reader unfamiliar with rat use.
-
Dr. Behl explained that the rats were positive for pinworms for the duration of the study, and they did not receive medication for elimination. At study termination, the incidences of pinworms were similar between the exposed groups. Based on histological sections, the pinworms were not associated with morphological changes in the large intestine and no inflammatory response was noted. NTP can clarify that point.
-
Dr. Wise indicated it would be helpful to include that no medication was administered.
-
-
Dr. Wise recommended that NTP add a second parameter in the table when presenting comet assay results, since it is standard to show two different measures such as tail length, olive moment, or tail moment.
-
Dr. Stephanie Smith-Roe noted that NTP uses percent tail DNA, which is the OECD guideline for comet assay. Some comet assays report more than one measure, with those measures usually based on tail length. As there is variability in electrophoretic conditions that can influence tail length, NTP has found that percent tail DNA is a more reliable measure.
-
Dr. Ludewig commented that she was unsure of how to interpret the significant comet effects even at the lowest concentration given the lack of pathology in the liver and other published positive comet assay results in the liver and other tissues.
-
Dr. Smith-Roe replied that the results suggest that sodium tungstate may be capable of damaging DNA, but the liver was not a target organ for neoplastic effects.
-
-
G.2.3.3.3
-
Dr. Mahrt commented that the studies were well-designed, well-conducted, and the report was clear. However, he suggested NTP clarify whether the progression in rats from uterine atypical hyperplasia to adenocarcinomas (noted in Table 31) also included an increase in uterine adenomas.
-
Dr. Brix agreed that it was unclear and indicated that NTP will consider adding language regarding the progression. From experience, adenomas are less common and NTP often sees a direct progression from atypical hyperplasia to adenocarcinomas.
-
G.2.3.3.4
Dr. Daniel Spade indicated he had no additional comments that were not already addressed by the other reviewers.
Dr. Ludewig noted that the comet assay should be brought forward into the abstract since DNA damage was observed in the liver, despite no associated pathology.
Dr. Tracie Bunton asked about the justification for the large number of interim sacrifices and questioned whether NTP could have obtained information on tungsten accumulation using fewer sacrifices.
-
Dr. Behl explained that the study was started several years ago when there were no data in the literature on the accumulation of tungsten in tissues following repeat dosing. Because the kidney is a target, NTP included multiple time points to determine if accumulation continued over the course of the study or if saturation eventually occurred. In addition, a question of sex differences required that NTP use males and females.
Dr. Bunton agreed with Dr. Mahrt’s comment about Table 31 and thought the discussion was sufficient. She asked why Table 31 did not include incidence and statistics.
-
Dr. Brix clarified that the adenocarcinomas and adenomas were included in Table 31 to show that the incidences were not significantly increased and that there was no progression from atypical hyperplasia. There were no statistics included because they were all negative.
Dr. Bunton noted that for female mice, there was a significant increase in the incidence of hepatocellular adenomas and carcinomas in the 500 mg/L group and that they were included in the “other tissues” groups. She asked why that was lumped into the “other tissues” category rather than brought into the tumor category.
-
Dr. Brix stated that hepatocellular tumors are a common background lesion in this strain of mice, so it is not uncommon to have a dose group with significant differences due to biological variation. However, there was no dose response or reason to consider these treatments related.
Dr. Bunton asked for additional language or reorganization to be added in the discussion to explain how NTP came to an equivocal conclusion for female rats. NTP stated that the conclusion was based on increased incidences of C-cell adenoma and carcinoma (combined) of the thyroid gland, but the statement is incomplete as written because that same rationale could apply for a carcinogenic conclusion.
-
Dr. Brix said that NTP could look at the discussion and clarify.
Dr. Ludewig asked if NTP had any information about adipose tissue or lipid content in the liver, given that absolute liver weight decreased, there were positive comet assay results in the males, and the text mentions that sodium tungstate dihydrate is an antidiabetic agent.
-
Dr. Nigel Walker said that NTP does not have these data.
G.2.3.4.
Vote on NTP Conclusions
Male Hsd:Sprague Dawley SD ratsG.2.3.4.1
Dr. Ludewig called for a motion from the panel to approve the conclusions as written. Dr. Wise so moved, and Dr. Mahrt seconded the motion. The panel voted unanimously (5 yes, 0 no, 0 abstentions) to approve the conclusions as written.
Female Hsd:Sprague Dawley SD ratsG.2.3.4.2
Dr. Ludewig called for a motion from the panel to approve the conclusions as written. Dr. Wise so moved, and Dr. Elwell seconded the motion. The panel voted unanimously (5 yes, 0 no, 0 abstentions) to approve the conclusions as written.
Male B6C3F1/N miceG.2.3.4.3
Dr. Ludewig called for a motion from the panel to approve the conclusions as written. Dr. Wise so moved, and Dr. Elwell seconded the motion. The panel voted unanimously (5 yes, 0 no, 0 abstentions) to approve the conclusions as written.
Female B6C3F1/N miceG.2.3.4.4
Dr. Ludewig called for a motion from the panel to approve the conclusions as written. Dr. Wise so moved, and Dr. Mahrt seconded the motion. The panel voted unanimously (5 yes, 0 no, 0 abstentions) to approve the conclusions as written.
G.2.4.
Toxicology and Carcinogenesis Studies of Di-n-butyl Phthalate
G.2.4.1.
Presentation and Clarifying Questions
Dr. Madelyn (Mimi) Huang summarized the studies and conclusions reported in the Draft NTP Technical Report on the Toxicology and Carcinogenesis Studies of Di-n-butyl Phthalate (CASRN 84-74-2) Administered in Feed to Sprague Dawley (Hsd:Sprague Dawley SD) Rats and B6C3F1/N Mice.
Di-n-butyl phthalate (DBP) is commonly used as a plasticizer and is found in a variety of consumer products, such as vinyl fabrics and flooring, personal care products, pharmaceuticals, and food packaging. Human exposure primarily occurs through ingestion of food packaged in materials containing DBP; some inhalation and dermal exposure occurs as well, but to a lesser extent. In the gut, DBP is rapidly metabolized to mono-n-butyl phthalate (MBP) and undergoes broad distribution throughout the body.
Dr. Huang presented a summary of results from the perinatal and postweaning toxicity and carcinogenicity study in Hsd:Sprague Dawley SD rats. Time-mated female rats were fed diets containing 0, 300, 1,000, 3,000, or 10,000 ppm DBP from gestation day (GD) 6 through postnatal day (PND) 21. NTP provided F1 generation rats with the same respective DBP concentrations in feed as their dam for 2 years (generally two/sex/litter). In addition, select dams and their litters were removed on GD 18 and lactation day (LD) 4 to quantify the internal concentration of MBP.
Dr. Huang then presented a summary of results from the chronic toxicity and carcinogenicity study in B6C3F1/N mice. Mice were fed diets containing 0, 1,000, 3,000, or 10,000 ppm DBP for 2 years (n = 50/sex/group).
Under the conditions of these 2-year studies, NTP’s draft conclusions were:
-
Equivocal evidence of carcinogenic activity in male Hsd:Sprague Dawley SD rats based on marginal increases in the incidence of pancreatic acinus adenomas.
-
Exposure to DBP resulted in increased incidences of gross lesions of the male reproductive system and nonneoplastic lesions of the male reproductive system, liver, and pituitary gland pars distalis in male rats.
-
No evidence of carcinogenic activity in female Hsd:Sprague Dawley SD rats at 300, 1,000, 3,000, or 10,000 ppm.
-
Exposure to DBP resulted in increased incidences of nonneoplastic lesions of the liver in female rats.
-
No evidence of carcinogenic activity in male B6C3F1/N mice at 1,000, 3,000, or 10,000 ppm.
-
Exposure to DBP increased incidences of nonneoplastic lesions of the male reproductive system and liver in male mice.
-
No evidence of carcinogenic activity in female B6C3F1/N mice at 1,000, 3,000, or 10,000 ppm.
-
Exposure to DBP increased incidences of nonneoplastic lesions of the liver and kidney in female mice.
There were no clarifying questions or comments about the presentation.
G.2.4.2.
Public Comments
Dr. Gabriele Ludewig acknowledged that there were no written public comments on the draft technical report. She also noted that the panel did not receive requests for oral public comments on the draft technical report.
G.2.4.3.
Peer-Review Comments and Panel Discussion
First Reviewer – Dr. Tracie BuntonG.2.4.3.1
-
Dr. Bunton said the presentation of the rationale, methods, and results was clear and concise.
-
She commented that she liked the introduction and found it helpful in getting up to speed on the properties, uses, and reduction in use of DBP. The rationale for the overall significance of the study is solid, especially given that perinatal exposure is a knowledge gap.
-
She indicated that all the information about stability, homogeneity, dose selection, and number of animals per dose group is appropriate for the design of the study.
-
Dr. Bunton suggested including the word “microscopic” or eliminate the word “gross” in the conclusion statement of the report as it currently specifies gross lesions in the male reproductive system but does not specify that the nonneoplastic lesions are not gross lesions.
-
Dr. Huang indicated that NTP is open to adding in the word “microscopic” to differentiate from gross lesions.
-
-
Dr. Bunton noted that the findings to support the “equivocal” decision included an increased incidence in pancreatic acinar adenomas compared to controls, without a concurrent increase in hyperplasia, and a significant positive trend.
-
She stated the lesion in the liver was compatible with effects of other phthalates, namely increased cytoplasmic alteration. This was an important point that was noted in the pathology review and in the discussion.
-
She also agreed that the findings fit into the “equivocal” category.
G.2.4.3.2
-
Dr. Mahrt agreed with Dr. Bunton’s comments and noted that the report was well-designed, conducted, and written.
-
He appreciated the references to the literature to put the findings in perspective, especially the possibility of lesions related to peroxisome proliferation.
G.2.4.3.3
-
Dr. Spade indicated that the study was well-conducted, and the report was clearly written.
-
He noted that in the abstract, lines 39 to 40 indicate that there were fewer and less severe reproductive lesions in mice than in rats. He agreed with that conclusion, though suggested qualifying it by acknowledging the limits of cross-species comparison given that rats had perinatal exposure and mice did not.
-
Dr. Huang agreed that it was important to qualify the rat versus mouse comparison and said NTP could clarify those statements.
-
Dr. Nigel Walker explained that when NTP started adding the perinatal component to rat studies, people were trying to understand why the study design changed. With rat studies as perinatal and mouse studies as adults only, NTP has started to address that in the mid-2000s. In addition, it is difficult to do perinatal exposure on F1 generation B6C3F1/N mice.
-
-
Dr. Spade built on Dr. Bunton’s comment about the equivocal conclusion for pancreatic acinar adenomas and asked if there was a significant trend test but not pairwise test.
-
Dr. Huang stated the affirmative.
-
Dr. Spade appreciated the response and agreed with the conclusion.
-
-
-
He also asked why images were not included in some cases.
-
Dr. Huang clarified that NTP does not generally include images for common lesions or when the conclusion is equivocal.
-
Dr. Spade appreciated the response and agreed with the approach.
-
-
G.2.4.3.4
Dr. Michael Elwell noted that the last line of the discussion mentions that 2,4-dichlorophenoxyacetic acid and DBP did not produce hepatic lesions typical of peroxisome proliferators. DBP did result in hepatic lesions but did not produce hepatic neoplasms.
-
Dr. Huang agreed that the last line should reference neoplasms instead of lesions, as Dr. Elwell suggested.
Regarding voting, Dr. Elwell noted that the original draft report mentions hypertrophy in the pituitary and hyperplasia, but that it was not mentioned in the oral presentation. He asked if hypertrophy was included in the pituitary findings.
-
Dr. Mark Cesta explained that NTP relooked at the data about the pituitary and hyperplasia. Hyperplasia often occurs with hypertrophy lesions, so they wanted to explain the observed hyperplasia. However, after a closer look, NTP could not make that conclusion and decided to remove it from the abstract.
Dr. Ludewig noted that page 78 of the draft report explains the concentrations found in the amniotic fluid. When humans and rats both take up 1 mg/kg/day, a historic report showed that there were 22 ng/mL of the metabolite in the amniotic fluid of humans. However, NTP only found 5 ng/mL in rats. Dr. Ludewig said she was initially surprised until she noticed that NTP did not measure the glucuronic acid conjugate. She wondered if that would also cross the placenta or if conjugation would protect the fetus. She suggested adding a statement that the gluconate conjugate was not measured.
-
Dr. Huang agreed that there are differences in distribution and metabolism between rodents and humans. She was unsure if glucuronidation affects its ability to cross the placenta and said that was something NTP would look into.
G.2.4.4.
Vote on NTP Conclusions
Male Hsd:Sprague Dawley SD ratsG.2.4.4.1
Dr. Ludewig called for a motion from the panel to approve the conclusions as written. Dr. Bunton so moved, and Dr. Mahrt seconded the motion. The panel voted unanimously (5 yes, 0 no, 0 abstentions) to approve the conclusions as written.
Female Hsd:Sprague Dawley SD ratsG.2.4.4.2
Dr. Ludewig called for a motion from the panel to approve the conclusions as written. Dr. John Pierce Wise so moved, and Dr. Elwell seconded the motion. The panel voted unanimously (5 yes, 0 no, 0 abstentions) to approve the conclusions as written.
Male B6C3F1/N miceG.2.4.4.3
Dr. Ludewig called for a motion from the panel to approve the conclusions as written. Dr. Bunton so moved, and Dr. Wise seconded the motion. The panel voted unanimously (5 yes, 0 no, 0 abstentions) to approve the conclusions as written.
Female B6C3F1/N miceG.2.4.4.4
Dr. Ludewig called for a motion from the panel to approve the conclusions as written. Dr. Wise so moved, and Dr. Bunton seconded the motion. The panel voted unanimously (5 yes, 0 no, 0 abstentions) to approve the conclusions as written.
G.2.5.
Toxicology and Carcinogenesis Studies of Di(2-ethylhexyl) Phthalate
G.2.5.1.
Presentation and Clarifying Questions
Dr. Chad Blystone summarized the studies and conclusions reported in the Draft NTP Technical Report on the Toxicology and Carcinogenesis Studies of Di(2-ethylhexyl) Phthalate (CASRN 117-81-7) Administered in Feed to Sprague Dawley (Hsd:Sprague Dawley SD) Rats.
Di(2-ethylhexyl) phthalate (DEHP) is a phthalate ester that was widely used in manufacturing of PVC polymers and corresponding products, such as cosmetics and toys. Over the years DEHP use has declined due to toxicity concerns, but chronic exposure throughout multiple life stages still occurs. The literature suggests that exposure to DEHP during an early life stage may result in chronic or carcinogenic health outcomes. However, previous DEHP chronic rodent studies did not include exposure during the gestational period up to weaning in rodents. To address this, NTP conducted two comparative DEHP carcinogenesis studies in rats to determine if including early life exposure would alter chronic toxicity or carcinogenicity outcomes.
Dr. Blystone presented a summary of results from the perinatal and postweaning toxicity/carcinogenicity study in Hsd:Sprague Dawley SD rats. In the perinatal and postweaning study, time-mated female rats were fed diets containing 0, 300, 1,000, 3,000, or 10,000 ppm DEHP from gestational day (GD) 6 through postnatal day (PND) 21 (n = 45/group). Select dams were removed on GD 18 to quantify internal concentrations of a metabolite of DEHP, mono(2-ethylhexyl) phthalate, in plasma and tissue samples. NTP provided F1 generation rats with the same respective DEHP concentration in feed as their dam for 2 years (n = 2/sex/litter; n = 50 total/sex/group).
Dr. Blystone then presented a summary of results from the postweaning toxicity/carcinogenicity study in Hsd:Sprague Dawley SD rats. In the postweaning-only study, rats were fed diets containing 0, 300, 1,000, 3,000, or 10,000 ppm DEHP for 2 years (n = 50/sex/group).
Under the conditions of these 2-year studies, NTP’s draft conclusions were:
-
Perinatal and Postweaning Feed Study:
-
Clear evidence of carcinogenic activity in male Hsd:Sprague Dawley SD rats based on the increased incidences of hepatocellular adenoma or carcinoma (combined) and acinar adenoma or carcinoma (combined) neoplasms (predominantly adenomas) of the pancreas.
-
Exposure to DEHP resulted in increased incidences of nonneoplastic lesions in the liver, heart, pituitary gland, testis, and epididymis and increased incidences of gross lesions of the reproductive tract, bone marrow, and kidney in male rats.
-
Clear evidence of carcinogenic activity in female Hsd:Sprague Dawley SD rats based on the increased incidence of hepatocellular adenoma or carcinoma (combined).
-
The occurrence of pancreatic acinar adenoma or carcinoma (combined) was considered to be related to exposure. (Some evidence)
-
The occurrence of uterine (including cervix) adenoma, adenocarcinoma, squamous cell carcinoma, or squamous cell papilloma (combined) in female rats may have been related to exposure. (Equivocal evidence)
-
Exposure to DEHP resulted in increased incidences of nonneoplastic lesions in the liver and increased incidences of gross lesions of the kidney in female rats.
-
-
Postweaning-only Feed Study
-
Clear evidence of carcinogenic activity in male Hsd:Sprague Dawley SD rats based on the increased incidences of hepatocellular adenoma or carcinoma (combined) and increased incidences of acinar adenoma or carcinoma (combined) neoplasms (predominantly adenomas) of the pancreas.
-
The occurrence of testicular interstitial cell adenoma in male rats may have been related to exposure. (Equivocal evidence)
-
Exposure to DEHP resulted in increased incidences of nonneoplastic lesions in the liver, pancreas, bone marrow, heart, pituitary gland, testis, and epididymis.
-
Clear evidence of carcinogenic activity in female Hsd:Sprague Dawley SD rats based on the incidences of hepatocellular adenoma or carcinoma (combined) and uterine (including cervix) adenoma, adenocarcinoma, squamous cell carcinoma, or squamous cell papilloma (combined).
-
The occurrence of pancreatic acinar adenoma or carcinoma (combined) in female rats was considered to be related to exposure. (Some evidence)
-
Exposure to DEHP resulted in increased incidences of nonneoplastic lesions in the liver, pancreas, bone marrow, and uterus in female rats.
-
-
Comparative Carcinogenic Benchmark Dose Analyses
-
No consistent pattern indicating that perinatal and postweaning exposure was more sensitive compared to postweaning-only exposure and modeled responses were within threefold of each other.
-
However, there was a stronger carcinogenic response in the reproductive organs (uterus and testis) in the postweaning-only exposure study compared to the perinatal and postweaning exposure study.
-
Dr. Gabriele Ludewig asked a clarifying question about a shift in male to female fetus ratios that was not mentioned during Dr. Blystone’s talk. Dr. Blystone responded that female fetuses were lost at the highest concentration. The reduction in litter size was due to this but was inconsistent and not considered related to DEHP exposure.
G.2.5.2.
Public Comments
Dr. Ludewig acknowledged that there were no written public comments submitted on the draft technical report. She noted that the panel also did not receive requests for oral public comments on the draft technical report.
G.2.5.3.
Peer-Review Comments and Panel Discussion
First Reviewer – Dr. John Pierce WiseG.2.5.3.1
-
Dr. Wise indicated that the study was well-designed, well-conducted, and clear in its data presentation.
-
He suggested that NTP clarify that pinworm infections were not treated with medication.
-
Dr. Blystone said that NTP can add this clarification.
-
-
Dr. Wise also reiterated his comment from sodium tungstate dihydrate that NTP should explain that the choice of dose was deliberate and not intended to test the most environmentally relevant level of exposure, but rather to test whether the substance is carcinogenic.
-
Dr. Blystone agreed that the point of these studies is hazard characterization and noted that NTP can clarify that in the report and include it in the lay summary.
-
Dr. Wise suggested that NTP could add this in a dose selection rationale section.
-
Dr. Blystone responded that the reports have a section covering the technical aspects of the exposure concentration selection and that a sentence can be added to highlight rationale for exposure concentrations.
-
G.2.5.3.2
-
Dr. Spade stated that the report was thorough and clear; it makes a massive amount of work easily understandable.
-
He asked for clarification in how the reduction in litter size was presented, and asked whether there was an effort to address post-implantation loss as part of the reduced litter size. Reduced litter size could not be due to exposure related pre-implantation loss since the dosing window did not begin before implantation. However, Dr. Spade argued that with exposures beginning on GD 6, this overlaps with organogenesis and poses a risk for post-implantation loss which has been reported in the phthalate literature.
-
Dr. Blystone said that NTP only evaluated post-implantation loss in females which did not deliver, so there are no more data available on post-implantation loss and litter size.
-
-
Dr. Spade asked why pup survival data for PND 1–4 and PND 5–21 on page 29 of the draft report were analyzed separately. Dr. Spade thought that there could have been a trend in mortality if PND 1–21 were analyzed together.
-
Dr. Blystone explained that NTP typically standardizes the litter size on PND 4, so the analysis looks at early (PND 1–4) and later (PND 5–21) mortality. Dr. Blystone acknowledged that based on Dr. Spade’s written comment NTP reanalyzed the data after combining the two periods and there was still no significant trend or pairwise comparison.
-
-
Dr. Spade also noted that it was unclear why some lesions of unknown biological significance on page 61 of the draft report were classified as such. Of note were the adrenal gland lesions, because of the known antiandrogenic effect of DEHP. Also, he questioned the ovarian atrophy as classified as unknown biological significance, because published data from academic studies indicate that phthalates change the rates of follicle maturation which could be related.
-
Dr. Blystone indicated that NTP can clarify the language and focus more on toxicological significance.
-
-
Dr. Spade commented that 24 months is reproductively aged, so for certain findings such as seminiferous epithelium degeneration, the control levels are very high which makes it less likely that there will be a significant pairwise test. This limits the ability to know with certainty what the dose response would look like for an endpoint such as epithelial degeneration. If 4-month-old males were tested, you might see a significant response at lower levels. The study supports the conclusions within the constraints of the study, but this is a limitation.
-
Dr. Blystone agreed that at this age, the model is not very sensitive. NTP can add a statement about that to the report.
-
-
Dr. Spade noted that for gestational transfer, as discussed on page 33 of the draft report, one limitation is that DEHP has many secondary metabolites. He noted that without measuring these secondary metabolites it is difficult to determine the total transfer.
-
Dr. Blystone agreed and said that NTP can clarify that metabolites can be transferred at different rates.
-
Dr. Ludewig stated that it was interesting that mono(2-ethylhexyl) phthalate was found in the amniotic fluid and the fetus of the control animals, but nothing in the serum of dams.
-
Dr. Blystone agreed that this was unusual and noted that sometimes sample preparations can lead to irregularities.
-
Dr. Suramya Waidyanatha said NTP concluded it was probably due to contamination of samples during collection or preparation and that this is likely due to the small volume of these samples.
G.2.5.3.3
-
Dr. Elwell agreed with previous reviewers that the results are clearly presented and discussed.
-
He noted that at the highest dose, body weights were reduced by approximately 30% in the perinatal postweaning and approximately 20% in the postweaning study. Although this did not affect survival, the decreased incidence of several neoplastic and nonneoplastic findings (especially for the high dose group of each study) might be attributable to significantly lower body weights. In the report (pages 61 and 77) these are indicated to be of unknown biological significance. If these findings are due to lower body weight, they should be addressed as such rather than reported as unknown significance. For example, neoplasms including the c-cell, pituitary, and mammary gland tumors can be affected by body weight. In females, a single pituitary gland neoplasm reported at the high dose is unexpected and notable in comparison to the control group where 16 animals were reported to have this tumor. The decreased incidence of the nonneoplastic lesions of testis polyarteritis and parathyroid hyperplasia could also be related to the lowered severity of chronic progressive nephropathy (CPN). In both studies, CPN was decreased in severity at the high dose in both studies and indicated as the cause of death in 0 and 1 animal while 16–18 animals in the control groups list CPN as the cause of death. This report cites other DEHP studies that attribute an increase in CPN to DEHP while this report suggests the opposite effect on CPN. This may be because the highest doses in this study were lower, the decreased body weight, or that the NTP 2000 diet was not used in earlier studies.
-
Dr. Blystone indicated that NTP can clarify biological significance versus toxicological significance. Dr. Blystone agreed that some of the effects could be related to decreased body weight and stated that NTP can clarify.
-
-
Dr. Elwell asked if page 87 of the draft report should state that it is unclear if any difference corresponded to developmental mechanisms when comparing the two studies, or if it is a typo. It appeared the kidney was affected by a developmental mechanism and DEHP was presumed to interfere with proper development.
-
Dr. Blystone stated that NTP can fix the typo.
-
-
Dr. Elwell noted that for the nonneoplastic conclusions, there are dozens of neoplastic and nonneoplastic findings clearly related to DEHP. He questioned why the acute inflammation in the uterus in the perinatal/postnatal study was considered an effect. He also asked why the bone marrow in females in the postweaning-only study was included as a finding and if it was possibly a false positive.
-
Dr. Blystone said that although the response was not strong, NTP considered the bone marrow lesion exposure related since it was observed in males and females. The acute inflammation in the uterus was considered exposure related. NTP can review this and clarify.
-
Dr. Susan Elmore stated that she can see Dr. Elwell’s point about the acute inflammation of the uterus possibly not being related to exposure, but that this was something NTP would need to discuss further.
-
G.2.5.3.4
Dr. Tracie Bunton said she wanted to see the presentation of the benchmark dose analysis, but beyond that, just had minor edits submitted in writing.
Dr. Spade asked if Dr. Elwell’s comments about bone marrow and uterus related to one of the conclusions on which the panel would vote. He wondered if it was about including it in the abstract rather than the conclusion that there was a finding.
-
Dr. Elwell said while the other findings listed in the abstract were convincing effects, the rationale to include the non-dose-related difference of acute uterus inflammation in the perinatal-post weaning study was not clear although there may have been reason to include it as chronic uterus inflammation was increased in the postweaning-only study. The question on including the bone marrow finding in females from the postweaning-only study in the abstract was based on very small group differences in incidence with no apparent effect on severity.
Dr. Bunton noted that a number of genetic toxicity tests were conducted and asked if they were related to this particular report or conducted over time.
-
Dr. Blystone said they were not related to this report itself. They were accumulated over time and not published previously, so they were published with this report.
G.2.5.4.
Vote on NTP Conclusions
Male Hsd:Sprague Dawley SD rats (perinatal and postweaning feed study)G.2.5.4.1
Dr. Ludewig called for a motion from the panel to approve the conclusions as written. Dr. Bunton so moved, and Dr. Wise seconded the motion. The panel voted unanimously (5 yes, 0 no, 0 abstentions) to approve the conclusions as written.
Female Hsd:Sprague Dawley SD rats (perinatal and postweaning feed study)G.2.5.4.2
Dr. Ludewig called for a motion from the panel to approve the conclusions as written. Dr. Wise so moved, and Dr. Elwell seconded the motion. Dr. Elwell asked to amend the motion to vote and moved that NTP delete “and increased incidences of gross lesions of the” from the conclusion and add “and uterus.” Dr. Wise motioned to accepted revisions to the conclusion and Dr. Spade seconded the motion. The panel voted unanimously (5 yes, 0 no, 0 abstentions) to approve the new conclusion.
In a second round of revisions, Dr. Blystone noted that the end of a sentence was cut off. He added “and gross observations in the female reproductive tract” to the end of the final conclusion. Dr. Ludewig called for a motion from the panel to approve the second round of revised conclusions. Dr. Wise so moved, and Dr. Elwell seconded the motion. The panel voted unanimously (5 yes, 0 no, 0 abstentions) to approve the new conclusion, below.
Revised Conclusion:
-
Clear evidence of carcinogenic activity
-
Increased incidence of hepatocellular adenoma or carcinoma (combined).
-
-
The occurrence of pancreatic acinar adenoma or carcinoma (combined) was considered to be related to exposure. (Some evidence)
-
The occurrence of uterine (including cervix) adenoma, adenocarcinoma, squamous cell carcinoma, or squamous cell papilloma (combined) in female rats may have been related to exposure. (Equivocal evidence)
-
Exposure to DEHP resulted in increased incidences of nonneoplastic lesions in the liver, and increased incidences of gross lesions of the kidney, and uterus in female rats and gross observations in the female reproductive tract.
G.2.5.4.3
Dr. Ludewig called for a motion from the panel to approve the conclusions as written. Dr. Wise so moved, and Dr. Elwell seconded the motion. The panel voted unanimously (5 yes, 0 no, 0 abstentions) to approve the conclusions as written.
Female Hsd:Sprague Dawley SD rats (postweaning-only study)G.2.5.4.4
Dr. Ludewig called for a motion from the panel to approve the conclusions as written. The panel did not offer a motion. Dr. Elwell moved that NTP delete the reference to increased incidences of nonneoplastic lesions in the bone marrow from the conclusion. Dr. Wise seconded the motion. The panel voted unanimously (5 yes, 0 no, 0 abstentions) to approve the new conclusion, below.
Revised Conclusion:
-
Clear evidence of carcinogenic activity
-
Increased incidences of hepatocellular adenoma or carcinoma (combined) and uterine (including cervix) adenoma, adenocarcinoma, squamous cell carcinoma, or squamous cell papilloma (combined).
-
-
The occurrence of pancreatic acinar adenoma or carcinoma (combined) in female rats was considered to be related to exposure. (Some evidence)
-
Exposure to DEHP resulted in increased incidences of nonneoplastic lesions in the liver, pancreas, bone marrow, and uterus in female rats.
G.2.5.4.5
Dr. Ludewig called for a motion from the panel to approve the conclusions as written. Dr. Wise so moved, and Dr. Elwell seconded the motion. The panel voted unanimously (5 yes, 0 no, 0 abstentions) to approve the conclusions as written.
G.2.5.5.
Final Conclusions
Because revisions were proposed and approved during the meeting, the final approved conclusions are presented below:
-
Perinatal and Postweaning Feed Study:
-
Clear evidence of carcinogenic activity in male Hsd:Sprague Dawley SD rats based on the increased incidences of hepatocellular adenoma or carcinoma (combined) and acinar adenoma or carcinoma (combined) neoplasms (predominantly adenomas) of the pancreas.
-
Exposure to DEHP resulted in increased incidences of nonneoplastic lesions in the liver, kidney, bone marrow, heart, pituitary gland, testis, and epididymis and increased incidences of gross lesions of the reproductive tract
-
Clear evidence of carcinogenic activity in female Hsd:Sprague Dawley SD rats based on the increased incidence of hepatocellular adenoma or carcinoma (combined).
-
The occurrence of pancreatic acinar adenoma or carcinoma (combined) was considered to be related to exposure. (Some evidence)
-
The occurrence of uterine (including cervix) adenoma, adenocarcinoma, squamous cell carcinoma, or squamous cell papilloma (combined) in female rats may have been related to exposure. (Equivocal evidence)
-
Exposure to DEHP resulted in increased incidences of nonneoplastic lesions in the liver, kidney, and uterus in female rats and gross observations in the female reproductive tract.
-
-
Postweaning-only Feed Study
-
Clear evidence of carcinogenic activity in male Hsd:Sprague Dawley SD rats based on the increased incidences of hepatocellular adenoma or carcinoma (combined) and increased incidences of acinar adenoma or carcinoma (combined) neoplasms (predominantly adenomas) of the pancreas.
-
The occurrence of testicular interstitial cell adenoma in male rats may have been related to exposure. (Equivocal evidence)
-
Exposure to DEHP resulted in increased incidences of nonneoplastic lesions in the liver, pancreas, bone marrow, heart, pituitary gland, testis, and epididymis.
-
Clear evidence of carcinogenic activity in female Hsd:Sprague Dawley SD rats based on the increased incidences of hepatocellular adenoma or carcinoma (combined) and uterine (including cervix) adenoma, adenocarcinoma, squamous cell carcinoma, or squamous cell papilloma (combined).
-
The occurrence of pancreatic acinar adenoma or carcinoma (combined) in female rats was considered to be related to exposure. (Some evidence)
-
Exposure to DEHP resulted in increased incidences of nonneoplastic lesions in the liver, pancreas, and uterus in female rats.
-
-
Comparative Carcinogenic Benchmark Dose Analyses
-
No consistent pattern indicating that perinatal and postweaning exposure was more sensitive compared to postweaning-only exposure and modeled responses were within threefold of each other.
-
However, there was a stronger carcinogenic response in the reproductive organs (uterus and testis) in the postweaning-only exposure study compared to the perinatal and postweaning exposure study.
-
G.2.6.
Closing Remarks on the Draft Reports
Dr. Gabriele Ludewig welcomed additional panel comments on the draft report. There were no additional comments.
Closing the meeting, Dr. Sheena Scruggs thanked all the peer-review panelists.
Dr. Ludewig added her thanks to the NTP staff and the panel members for their efforts.
Dr. Ludewig adjourned the meeting at 2:00 p.m. EDT on April 2, 2021.
Appendix H. Supplemental Data
Tables with supplemental data can be found here: https://doi.org/10.22427/NTP-DATA-TR-601.202
About This Report
Foreword
The National Toxicology Program (NTP), established in 1978, is an interagency program within the Public Health Service of the U.S. Department of Health and Human Services. Its activities are executed through a partnership of the National Institute for Occupational Safety and Health (part of the Centers for Disease Control and Prevention), the Food and Drug Administration (primarily at the National Center for Toxicological Research), and the National Institute of Environmental Health Sciences (part of the National Institutes of Health), where the program is administratively located. NTP offers a unique venue for the testing, research, and analysis of agents of concern to identify toxic and biological effects, provide information that strengthens the science base, and inform decisions by health regulatory and research agencies to safeguard public health. NTP also works to develop and apply new and improved methods and approaches that advance toxicology and better assess health effects from environmental exposures.
The Technical Report series began in 1976 with carcinogenesis studies conducted by the National Cancer Institute. In 1981, this bioassay program was transferred to NTP. The studies described in the NTP Technical Report series are designed and conducted to characterize and evaluate the toxicological potential, including carcinogenic activity, of selected substances in laboratory animals (usually two species, rats and mice). Substances (e.g., chemicals, physical agents, and mixtures) selected for NTP toxicity and carcinogenicity studies are chosen primarily on the basis of human exposure, level of commercial production, and chemical structure. The interpretive conclusions presented in NTP Technical Reports are derived solely from the results of these NTP studies, and extrapolation of the results to other species, including characterization of hazards and risks to humans, requires analyses beyond the intent of these reports. Selection for study per se is not an indicator of a substance’s carcinogenic potential.
NTP conducts its studies in compliance with its laboratory health and safety guidelines and the Food and Drug Administration Good Laboratory Practice Regulations and meets or exceeds all applicable federal, state, and local health and safety regulations. Animal care and use are in accordance with the Public Health Service Policy on Humane Care and Use of Laboratory Animals. Studies are subjected to retrospective quality assurance audits before they are presented for public review. Draft reports undergo external peer review before they are finalized and published.
The NTP Technical Reports are available free of charge on the NTP website and cataloged in PubMed, a free resource developed and maintained by the National Library of Medicine (part of the National Institutes of Health). Data for these studies are included in NTP’s Chemical Effects in Biological Systems database.
For questions about the reports and studies, please email NTP or call 984-287-3211.
Collaborators
Division of the National Toxicology Program, National Institute of Environmental Health Sciences, Research Triangle Park, North Carolina, USA
Designed studies, evaluated and interpreted results, and reported findings
C.R. Blystone, Ph.D., Co-Study Scientist
T.D. Hubbard, Ph.D., Co-Study Scientist
S.A. Elmore, D.V.M., M.S., Study Pathologist
M.F. Cesta, D.V.M., Ph.D.
M.C. Cora, D.V.M.
H.C. Cunny, Ph.D.
P.M. Foster, Ph.D. (Retired)
M.J. Hooth, Ph.D.
A.P. King-Herbert, D.V.M.
D.E. Malarkey, D.V.M., Ph.D. (Retired)
B.S. McIntyre, Ph.D.
F.M. Parham, Ph.D.
G.K. Roberts, Ph.D.
V.G. Robinson, M.S.
K.A. Shipkowski, Ph.D.
K.R. Shockley, Ph.D.
M.D. Stout, Ph.D.
G.S. Travlos, D.V.M.
S. Waidyanatha, Ph.D.
N.J. Walker, Ph.D.
A.T.D. Watson, Ph.D.
K.L. Witt, M.S.
Provided oversight for data management
J.M. Fostel, Ph.D.
Battelle, Columbus, Ohio, USA
Conducted studies and evaluated pathology findings
M.R. Hejtmancik, Ph.D., Principal Investigator
C.A. Granville, Ph.D.
D.Y. Vasconcelos, D.V.M., Ph.D.
Conducted prestart chemistry activities and dose formulations
B.L. Burback, Ph.D.
D.A. Contos, M.S.
N.L. South, B.S.
RTI International, Research Triangle Park, North Carolina, USA
Conducted preliminary chemistry activities, dose formulations, and biological sample chemistry analyses
R.A. Fernando, Ph.D., Principal Investigator
J.C. Blake, B.A., Deputy Principal Investigator
S.D. Cooper, M.S.
J. Gilliam, B.S.
J. Lieause, M.S.
M. Silinski, Ph.D.
Experimental Pathology Laboratories, Inc., Research Triangle Park, North Carolina, USA
Provided pathology review
T.A. Crabbs, D.V.M.
M. Gruebbel, D.V.M., Ph.D.
G.A. Willson, B.V.M.S.
Coordinated NTP Pathology Working Groups on perinatal and postweaning rats (Study 1) (May 10, 2018) and on postweaning-only rats (Study 2) (September 29, 2016)
T.A. Crabbs, D.V.M. (Study 1)
G.A. Willson, B.V.M.S. (Study 2)
Coordinated NTP Pathology Peer Review on brain glial cell proliferative lesions in rats (Study 1 and Study 2) (April 18, 2016)
M. Gruebbel, D.V.M., Ph.D.
Coordinated NTP Pathology Peer Review on brain granular cell proliferative lesions and head/neck schwannomas/sarcomas in rats (Study 1 and Study 2) (April 18, 2016)
M. Gruebbel, D.V.M., Ph.D.
ASRC Federal, Research Triangle Park, North Carolina, USA
Prepared data for report
P. Brown, B.S.
H. Gong, M.S.
C. Martini, B.S.
C. Myers, M.S.
N. Sayers, B.S.
M. Shaw, B.S.
R. Whittlesey, M.S.
Contributors
Division of the National Toxicology Program, National Institute of Environmental Health Sciences, Research Triangle Park, North Carolina, USA
Provided oversight for external peer review
S.L. Scruggs, Ph.D.
M.S. Wolfe, Ph.D.
Kelly Government Services, Research Triangle Park, North Carolina, USA
Supported external peer review
E.A. Maull, Ph.D. (retired from NIEHS, Research Triangle Park, North Carolina, USA)
NTP Pathology Working Group, National Institute of Environmental Health Sciences, Research Triangle Park, North Carolina, USA
Participated in NTP Pathology Working Group on perinatal and postweaning rats (Study 1) and postweaning-only rats (Study 2) (August 14, 2018)
N. Barlow, D.V.M., Ph.D., Seattle Genetics, Inc.
M.F. Cesta, D.V.M., Ph.D., National Institute of Environmental Health Sciences
S.A. Elmore, D.V.M., M.S., National Institute of Environmental Health Sciences
R.A. Herbert, D.V.M., Ph.D., National Institute of Environmental Health Sciences
R.C. Kovi, M.V.Sc., Ph.D., Experimental Pathology Laboratories, Inc.
D.E. Malarkey, D.V.M., Ph.D., National Institute of Environmental Health Sciences
C.J. Willson, D.V.M., Ph.D., Integrated Laboratory Systems, LLC
NTP Pathology Peer Review, National Institute of Environmental Health Sciences, Research Triangle Park, North Carolina, USA
Participated in NTP Pathology Peer Review on control rat hearts (Study 1 and Study 2) (April 14, 2016)
M.F. Cesta, D.V.M., Ph.D., National Institute of Environmental Health Sciences
S.A. Elmore, D.V.M., M.S., National Institute of Environmental Health Sciences
M.P. Jokinen, D.V.M., Integrated Laboratory Systems, LLC
D.E. Malarkey, D.V.M., Ph.D., National Institute of Environmental Health Sciences
Participated in NTP Pathology Peer Review on brain glial cell proliferative lesions (Study 1 and Study 2) (April 18, 2016)
M.F. Cesta, D.V.M., Ph.D., National Institute of Environmental Health Sciences
P. Little, D.V.M., Ph.D., Experimental Pathology Laboratories, Inc.
D.E. Malarkey, D.V.M., Ph.D., National Institute of Environmental Health Sciences
Participated in NTP Pathology Peer Review on brain granular cell proliferative lesions and head/neck schwannomas and sarcomas (Study 1 and Study 2) (April 18, 2016)
M.F. Cesta, D.V.M., Ph.D., National Institute of Environmental Health Sciences
P. Little, D.V.M., Ph.D., Experimental Pathology Laboratories, Inc.
D.E. Malarkey, D.V.M., Ph.D., National Institute of Environmental Health Sciences
Performed special review of testis lesions (Study 1) (September 5, 2018)
K.Y. Cimon, D.V.M., Experimental Pathology Laboratories, Inc.
Experimental Pathology Laboratories, Inc., Research Triangle Park, North Carolina, USA
Supervised pathology review
T.J. Steinbach, D.V.M., Principal Investigator
G.A. Willson, B.V.M.S., Principal Investigator
Integrated Laboratory Systems, LLC, Research Triangle Park, North Carolina, USA
Conducted micronucleus assays
R.R. Tice, Ph.D., Principal Investigator
Brookhaven National Laboratory, Upton, New York, USA
Conducted chromosomal aberration and micronucleus assays
R.R. Tice, Ph.D., Principal Investigator
CSS Corporation, Research Triangle Park, North Carolina, USA
Prepared quality assessment audits
S. Brecher, Ph.D., Principal Investigator
S. Iyer, B.S.
V.S. Tharakan, D.V.M.
Social & Scientific Systems, a DLH Company, Research Triangle Park, North Carolina, USA
Provided statistical analyses
S.J. McBride, Ph.D., Principal Investigator
L.J. Betz, M.S.
S.F. Harris, M.S.
J.D. Krause, Ph.D.
ICF, Fairfax, Virginia, USA
Provided contract oversight
D. Burch, M.E.M., Principal Investigator
J. Cleland, M.E.M.
J.A. Wignall, M.S.P.H.
Prepared and edited report
K.S. Duke, Ph.D.
T. Hamilton, M.S.
B. Ingle, Ph.D.
K. McKinley, M.E.M.
M.E. McVey, Ph.D.
J.I. Powers, M.A.P.
K.E. Setty, Ph.D.
K.A. Shipkowski, Ph.D.
S.J. Snow, Ph.D.
J.W. Tracy, M.H.S.
Supported external peer review
C.N. Byrd, B.S.
L.M. Green, M.P.H.
M.C. Rooney, B.A.
A.J. Schumacher, B.A.
Explanation of Levels of Evidence of Carcinogenic Activity
The National Toxicology Program (NTP) describes the results of individual experiments on a chemical agent and notes the strength of the evidence for conclusions regarding each study. Negative results, in which the study animals do not have a greater incidence of neoplasia than control animals, do not necessarily mean that a chemical is not a carcinogen, in as much as the experiments are conducted under a limited set of conditions. Positive results demonstrate that a chemical is carcinogenic for laboratory animals under the conditions of the study and indicate that exposure to the chemical has the potential for hazard to humans. Other organizations, such as the International Agency for Research on Cancer, assign a strength of evidence for conclusions based on an examination of all available evidence, including animal studies such as those conducted by NTP, epidemiologic studies, and estimates of exposure. Thus, the actual determination of risk to humans from chemicals found to be carcinogenic in laboratory animals requires a wider analysis that extends beyond the purview of these studies.
Five categories of evidence of carcinogenic activity are used in the Technical Report series to summarize the strength of evidence observed in each experiment: two categories for positive results (clear evidence and some evidence); one category for uncertain findings (equivocal evidence); one category for no observable effects (no evidence); and one category for experiments that cannot be evaluated because of major flaws (inadequate study). These categories of interpretative conclusions were first adopted in June 1983 and then revised on March 1986 for use in the Technical Report series to incorporate more specifically the concept of actual weight of evidence of carcinogenic activity. For each separate experiment (male rats, female rats, male mice, female mice), one of the following five categories is selected to describe the findings. These categories refer to the strength of the experimental evidence and not to potency or mechanism.
-
Clear evidence of carcinogenic activity is demonstrated by studies that are interpreted as showing a dose-related (i) increase of malignant neoplasms, (ii) increase of a combination of malignant and benign neoplasms, or (iii) marked increase of benign neoplasms if there is an indication from this or other studies of the ability of such tumors to progress to malignancy.
-
Some evidence of carcinogenic activity is demonstrated by studies that are interpreted as showing a chemical-related increased incidence of neoplasms (malignant, benign, or combined) in which the strength of the response is less than that required for clear evidence.
-
Equivocal evidence of carcinogenic activity is demonstrated by studies that are interpreted as showing a marginal increase of neoplasms that may be chemical related.
-
No evidence of carcinogenic activity is demonstrated by studies that are interpreted as showing no chemical-related increases in malignant or benign neoplasms.
-
Inadequate study of carcinogenic activity is demonstrated by studies that, because of major qualitative or quantitative limitations, cannot be interpreted as valid for showing either the presence or absence of carcinogenic activity.
For studies showing multiple chemical-related neoplastic effects that if considered individually would be assigned to different levels of evidence categories, the following convention has been adopted to convey completely the study results. In a study with clear evidence of carcinogenic activity at some tissue sites, other responses that alone might be deemed some evidence are indicated as “were also related” to chemical exposure. In studies with clear or some evidence of carcinogenic activity, other responses that alone might be termed equivocal evidence are indicated as “may have been” related to chemical exposure.
When a conclusion statement for a particular experiment is selected, consideration must be given to key factors that would extend the actual boundary of an individual category of evidence. Such consideration should allow for incorporation of scientific experience and current understanding of long-term carcinogenesis studies in laboratory animals, especially for those evaluations that may be on the borderline between two adjacent levels. These considerations should include:
-
adequacy of the experimental design and conduct;
-
occurrence of common versus uncommon neoplasia;
-
progression (or lack thereof) from benign to malignant neoplasia as well as from preneoplastic to neoplastic lesions;
-
some benign neoplasms have the capacity to regress but others (of the same morphologic type) progress. At present, it is impossible to identify the difference. Therefore, where progression is known to be a possibility, the most prudent course is to assume that benign neoplasms of those types have the potential to become malignant;
-
combining benign and malignant tumor incidence known or thought to represent stages of progression in the same organ or tissue;
-
latency in tumor induction;
-
multiplicity in site-specific neoplasia;
-
metastases;
-
supporting information from proliferative lesions (hyperplasia) in the same site of neoplasia or other experiments (same lesion in another sex or species);
-
presence or absence of dose relationships;
-
statistical significance of the observed tumor increase;
-
concurrent control tumor incidence as well as the historical control rate and variability for a specific neoplasm;
-
survival-adjusted analyses and false positive or false negative concerns;
-
structure-activity correlations; and
-
in some cases, genetic toxicology.
Peer Review
The National Toxicology Program (NTP) convened a virtual external ad hoc panel to peer review the draft NTP Technical Report on the Toxicology and Carcinogenesis Studies of Di(2-ethylhexyl) Phthalate (CASRN 117-81-7) Administered in Feed to Sprague Dawley Hsd:Sprague Dawley SD) Rats on April 2, 2021. NTP announced the peer-review meeting in the Federal Register (86 FR. 9947. February 17, 2021). The public could view the proceedings online and opportunities were provided for submission of written and oral public comments. The selection of panel members and conduct of the peer review were in accordance with federal policies and regulations. The panel was charged to:
-
Review and evaluate the scientific and technical elements of each study and its presentation.
-
Determine whether each study’s experimental design, conduct, and findings support the NTP’s conclusions regarding the conditions of each study.
NTP carefully considered the panel’s recommendations in finalizing the report. The peer-review report is provided in Appendix G. Other meeting materials are available on the NTP website (https://ntp.niehs.nih.gov/go/meeting).
Peer Reviewers
Gabriele Ludewig, Ph.D., Chairperson
Professor, Department of Occupational and Environmental Health, College of Public Health
University of Iowa
Iowa City, Iowa, USA
Tracie Bunton, D.V.M., Ph.D.
Founder
Eicarte LLC: Pharmaceutical Preclinical Toxicology and Pathology Consultancy
Gettysburg, Pennsylvania, USA
Michael Elwell, D.V.M., Ph.D.
Consultant, Toxicologic Pathology
APEX TOXPATH, LLC
Apex, North Carolina, USA
Charles Mahrt, D.V.M., Ph.D.
Flagship Biosciences (Retired)
Broomfield, Colorado, USA
Daniel Spade, Ph.D.
Assistant Professor, Department of Pathology and Laboratory Medicine
Brown University
Providence, Rhode Island, USA
John Pierce Wise, Sr., Ph.D.
Professor, Department of Pharmacology and Toxicology
University of Louisville
Louisville, Kentucky, USA
Publication Details
Publisher: National Toxicology Program
Publishing Location: Research Triangle Park, NC
ISSN: 2378-8925
DOI: https://doi.org/10.22427/NTP-TR-601
Report Series: NTP Technical Report Series
Report Series Number: 601
Official citation: National Toxicology Program (NTP). 2021. NTP technical report on the toxicology and carcinogenesis studies of di(2-ethylhexyl) phthalate (CASRN 117-81-7) administered in feed to Sprague Dawley (Hsd:Sprague Dawley SD) rats. Research Triangle Park, NC: National Toxicology Program. Technical Report 601.
Acknowledgments
This work was supported by the Intramural Research Program (ES103316, ES103318, and ES103319) at the National Institute of Environmental Health Sciences, National Institutes of Health and performed for the National Toxicology Program, Public Health Service, U.S. Department of Health and Human Services under contracts HHSN273201800006C, HHSN271201800012I, GS00Q14OADU417 (Order No. HHSN273201600015U), HHSN273201600011C, HHSN273201500006C, HHSN273201500014C, HHSN273200800005C, HHSN273201400022C, HHSN273201300009C, HHSN316201200054W, HHSN273201100003C, N01-ES-1-75412, N01-ES-55541, and N01-ES-65554.
Overview
Phthalates are plasticizers used to provide flexibility in products composed of polyvinyl chloride plastic or vinyl chloride resins. Studies have shown that in utero and early life phthalate exposure can result in adverse reproductive, developmental, and potentially carcinogenic effects. The National Toxicology Program therefore initiated a broad-based program of work to provide toxicity data and a cancer hazard assessment for lifetime exposure to environmental phthalates. Data generated from this program are intended to facilitate cumulative and aggregate risk characterization efforts for multiple phthalates, including di(2-ethylhexyl) phthalate (DEHP), di-n-butyl phthalate, and di-isobutyl phthalate.
Previous carcinogenicity assessments of phthalates did not include perinatal exposure (gestation and lactation), whereas human exposure studies indicate that there is lifetime exposure to some phthalates. Thus, whether developmental exposure would alter lifetime DEHP-associated carcinogenic risk is unknown. Exposure during these critical periods of development and growth might be relevant for the evaluation of lifetime toxicological and carcinogenic risk.
The two 2-year toxicity and carcinogenicity studies described in this technical report were conducted to assess whether perinatal exposure would alter lifetime DEHP-associated carcinogenic risk. Specifically, the goal of the studies was to evaluate whether exposure to DEHP during the perinatal period would influence the pattern, dose response, incidence, or severity of the carcinogenic or noncarcinogenic response in rats relative to chronic exposure that does not include this critical period of development and growth.